
SAB2100872
Anti-GAA antibody produced in rabbit
affinity isolated antibody
Synonim(y):
Anti-Glucosidase, α; acid, Anti-LYAG
About This Item
Polecane produkty








pochodzenie biologiczne
rabbit
Poziom jakości
białko sprzężone
unconjugated
forma przeciwciała
affinity isolated antibody
rodzaj przeciwciała
primary antibodies
klon
polyclonal
Postać
buffered aqueous solution
masa cząsteczkowa
98 kDa
reaktywność gatunkowa
dog, guinea pig, human, rat, mouse, bovine
stężenie
0.5 mg - 1 mg/mL
metody
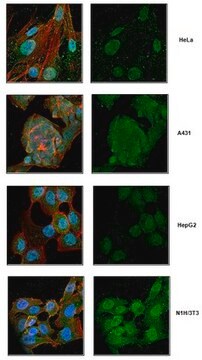
immunohistochemistry: suitable
western blot: suitable
numer dostępu UniProt
Warunki transportu
wet ice
temp. przechowywania
−20°C
docelowa modyfikacja potranslacyjna
unmodified
informacje o genach
human ... GAA(2548)
Opis ogólny
Immunogen
Zastosowanie
Działania biochem./fizjol.
Sekwencja
Postać fizyczna
Oświadczenie o zrzeczeniu się odpowiedzialności
Nie możesz znaleźć właściwego produktu?
Wypróbuj nasz Narzędzie selektora produktów.
Kod klasy składowania
10 - Combustible liquids
Klasa zagrożenia wodnego (WGK)
WGK 3
Temperatura zapłonu (°F)
Not applicable
Temperatura zapłonu (°C)
Not applicable
Certyfikaty analizy (CoA)
Poszukaj Certyfikaty analizy (CoA), wpisując numer partii/serii produktów. Numery serii i partii można znaleźć na etykiecie produktu po słowach „seria” lub „partia”.
Masz już ten produkt?
Dokumenty związane z niedawno zakupionymi produktami zostały zamieszczone w Bibliotece dokumentów.
Nasz zespół naukowców ma doświadczenie we wszystkich obszarach badań, w tym w naukach przyrodniczych, materiałoznawstwie, syntezie chemicznej, chromatografii, analityce i wielu innych dziedzinach.
Skontaktuj się z zespołem ds. pomocy technicznej