
WH0002548M1
Monoclonal Anti-GAA antibody produced in mouse
clone 3C6, purified immunoglobulin, buffered aqueous solution
Szinonimák:
Anti-LYAG, Anti-glucosidase, α acid (Pompe disease, glycogen storage disease type II)
About This Item
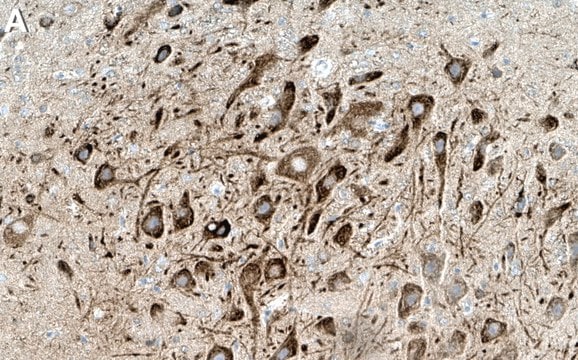
WB
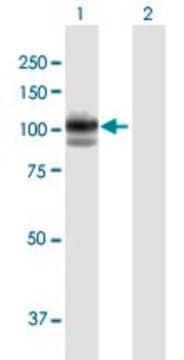
western blot: 1-5 μg/mL
Javasolt termékek





biológiai forrás
mouse
Minőségi szint
konjugátum
unconjugated
antitest forma
purified immunoglobulin
antitest terméktípus
primary antibodies
klón
3C6, monoclonal
Forma
buffered aqueous solution
faj reaktivitás
human
technika/technikák
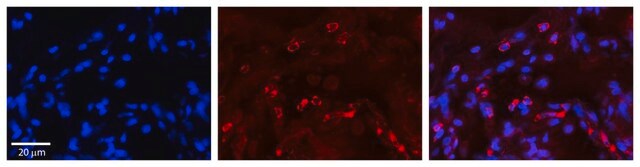
indirect ELISA: suitable
western blot: 1-5 μg/mL
izotípus
IgG1κ
GenBank elérési szám
UniProt elérési szám
kiszállítva
dry ice
tárolási hőmérséklet
−20°C
célzott transzláció utáni módosítás
unmodified
Géninformáció
human ... GAA(2548)
Általános leírás
Immunogén
Sequence
GEARGELFWDDGESLEVLERGAYTQVIFLARNNTIVNELVRVTSEGAGLQLQKVTVLGVATAPQQVLSNGVPVSNFTYSPDTKVLDICVSLLMGEQFLVSWC
Biokémiai/fiziológiai hatások
Fizikai forma
Jogi információk
Jogi nyilatkozat
Nem találja a megfelelő terméket?
Próbálja ki a Termékválasztó eszköz. eszközt
Tárolási osztály kódja
10 - Combustible liquids
Lobbanási pont (F)
Not applicable
Lobbanási pont (C)
Not applicable
Egyéni védőeszköz
Eyeshields, Gloves, multi-purpose combination respirator cartridge (US)
Válasszon a legfrissebb verziók közül:
Analitikai tanúsítványok (COA)
Nem találja a megfelelő verziót?
Ha egy adott verzióra van szüksége, a tétel- vagy cikkszám alapján rákereshet egy adott tanúsítványra.
Már rendelkezik ezzel a termékkel?
Az Ön által nemrégiben megvásárolt termékekre vonatkozó dokumentumokat a Dokumentumtárban találja.
Tudóscsoportunk valamennyi kutatási területen rendelkezik tapasztalattal, beleértve az élettudományt, az anyagtudományt, a kémiai szintézist, a kromatográfiát, az analitikát és még sok más területet.
Lépjen kapcsolatba a szaktanácsadással