
推薦產品








生物源
rabbit
品質等級
共軛
unconjugated
抗體表格
affinity isolated antibody
抗體產品種類
primary antibodies
無性繁殖
polyclonal
形狀
buffered aqueous solution
分子量
98 kDa
物種活性
dog, guinea pig, human, rat, mouse, bovine
濃度
0.5 mg - 1 mg/mL
技術
immunohistochemistry: suitable
western blot: suitable
UniProt登錄號
運輸包裝
wet ice
儲存溫度
−20°C
目標翻譯後修改
unmodified
基因資訊
human ... GAA(2548)
一般說明
Lysosomal alpha-glucosidase (GAA) is a glycoprotein that belongs to the glycoside hydrolase family GH31. It comprises a catalytic GH31 (β/α)8 barrel domain, trefoil type-P domain, an N-terminal β-sheet domain. The GAA gene is mapped to human chromosome location 17q25.3.
免疫原
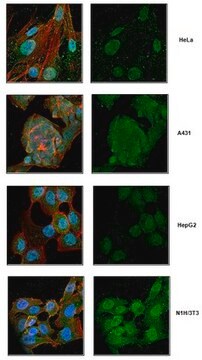
Synthetic peptide directed towards the N terminal region of human GAA
生化/生理作用
GAA is acid alpha-glucosidase, which is essential for the degradation of glycogen to glucose in lysosomes. Different forms of acid alpha-glucosidase are obtained by proteolytic processing. Defects in this gene are the cause of glycogen storage disease II, also known as Pompe′s disease, which is an autosomal recessive disorder with a broad clinical spectrum. This gene encodes acid alpha-glucosidase, which is essential for the degradation of glycogen to glucose in lysosomes. Different forms of acid alpha-glucosidase are obtained by proteolytic processing. Defects in this gene are the cause of glycogen storage disease II, also known as Pompe′s disease, which is an autosomal recessive disorder with a broad clinical spectrum. Three transcript variants encoding the same protein have been found for this gene.
序列
Synthetic peptide located within the following region: FGVIVRRQLDGRVLLNTTVAPLFFADQFLQLSTSLPSQYITGLAEHLSPL
外觀
Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and 2% sucrose.
免責聲明
Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to humans or animals.
未找到適合的產品?
試用我們的產品選擇工具.
儲存類別代碼
10 - Combustible liquids
水污染物質分類(WGK)
WGK 3
閃點(°F)
Not applicable
閃點(°C)
Not applicable
Cinzia Bragato et al.
Biochimica et biophysica acta. Molecular basis of disease, 1866(5), 165662-165662 (2020-01-10)
Pompe disease (PD) is an autosomal recessive muscular disorder caused by deficiency of the glycogen hydrolytic enzyme acid α-glucosidase (GAA). The enzyme replacement therapy, currently the only available therapy for PD patients, is efficacious in improving cardiomyopathy in the infantile
Hossein Moravej et al.
Iranian journal of medical sciences, 43(2), 218-222 (2018-05-12)
Pompe disease (PD), also known as "glycogen storage disease type II (OMIM # 232300)" is a rare autosomal recessive disorder characterized by progressive glycogen accumulation in cellular lysosomes. It ultimately leads to cellular damage. Infantile-onset Pompe disease (IOPD) is the
Francesco Chemello et al.
Cell reports, 26(13), 3784-3797 (2019-03-28)
Skeletal muscle is composed of different myofiber types that preferentially use glucose or lipids for ATP production. How fuel preference is regulated in these post-mitotic cells is largely unknown, making this issue a key question in the fields of muscle
Xiaodong Jia et al.
Aging, 12(5), 4268-4282 (2020-03-04)
Clinical manifestations of the late-onset adult Pompe disease (glycogen storage disease type II) are heterogeneous. To identify genetic defects of a special patient population with cerebrovascular involvement as the main symptom, we performed whole-genome sequencing (WGS) analysis on a consanguineous
Véronique Roig-Zamboni et al.
Nature communications, 8(1), 1111-1111 (2017-10-25)
Pompe disease, a rare lysosomal storage disease caused by deficiency of the lysosomal acid α-glucosidase (GAA), is characterized by glycogen accumulation, triggering severe secondary cellular damage and resulting in progressive motor handicap and premature death. Numerous disease-causing mutations in the
Active Filters
我們的科學家團隊在所有研究領域都有豐富的經驗,包括生命科學、材料科學、化學合成、色譜、分析等.
聯絡技術服務