
推薦產品










產品名稱
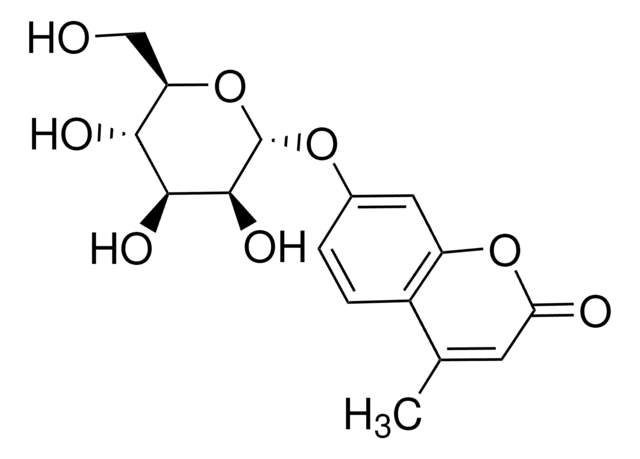
4-甲基伞形酮 α- D -吡喃葡萄糖苷, α-glucosidase substrate
描述
α-glucosidase substrate
化驗
≥99% (TLC)
形狀
powder
溶解度
pyridine: 50 mg/mL, clear, colorless to faintly yellow
儲存溫度
−20°C
SMILES 字串
CC1=CC(=O)Oc2cc(O[C@H]3O[C@H](CO)[C@@H](O)[C@H](O)[C@H]3O)ccc12
InChI
1S/C16H18O8/c1-7-4-12(18)23-10-5-8(2-3-9(7)10)22-16-15(21)14(20)13(19)11(6-17)24-16/h2-5,11,13-17,19-21H,6H2,1H3/t11-,13-,14+,15-,16+/m1/s1
InChI 密鑰
YUDPTGPSBJVHCN-JZYAIQKZSA-N
尋找類似的產品? 前往 產品比較指南
儲存類別代碼
11 - Combustible Solids
水污染物質分類(WGK)
WGK 3
閃點(°F)
Not applicable
閃點(°C)
Not applicable
個人防護裝備
Eyeshields, Gloves, type N95 (US)
從最近期的版本中選擇一個:
分析證明 (COA)
Lot/Batch Number
客戶也查看了




Omid Motabar et al.
Analytical biochemistry, 390(1), 79-84 (2009-04-18)
Mutations in alpha-glucosidase cause accumulation of glycogen in lysosomes, resulting in Pompe disease, a lysosomal storage disorder. Small molecule chaperones that bind to enzyme proteins and correct the misfolding and mistrafficking of mutant proteins have emerged as a new therapeutic
Intrapleural administration of AAV9 improves neural and cardiorespiratory function in Pompe disease.
Darin J Falk et al.
Molecular therapy : the journal of the American Society of Gene Therapy, 21(9), 1661-1667 (2013-06-05)
Pompe disease is a neuromuscular disease resulting from deficiency in acid α-glucosidase (GAA), results in cardiac, skeletal muscle, and central nervous system (CNS) pathology. Enzyme replacement therapy (ERT) has been shown to partially correct cardiac and skeletal muscle dysfunction. However
Phillip A Doerfler et al.
Human gene therapy, 27(1), 43-59 (2015-11-26)
Pompe disease is a progressive neuromuscular disorder caused by lysosomal accumulation of glycogen from a deficiency in acid alpha-glucosidase (GAA). Replacement of the missing enzyme is available by repeated protein infusions; however, efficacy is limited by immune response and inability
Ryoga Hamura et al.
Cancer science, 112(6), 2335-2348 (2021-05-02)
Lysosomal degradation plays a crucial role in the metabolism of biological macromolecules supplied by autophagy. The regulation of the autophagy-lysosome system, which contributes to intracellular homeostasis, chemoresistance, and tumor progression, has recently been revealed as a promising therapeutic approach for
Renata G K Leuschner et al.
Journal of AOAC International, 87(3), 604-613 (2004-08-04)
A standard method for the detection of Enterobacteriaceae was modified for the presumptive detection of Enterobacter sakazakii, and the modified method was validated in an interlaboratory trial with 16 laboratories from 8 European countries. The modification included a differential-elective medium
文章
Probiotics exhibit an inhibitory effect on pathogens, help prevent chronic intestinal inflammatory diseases or atopic syndromes, and support the immune system.
Active Filters
我們的科學家團隊在所有研究領域都有豐富的經驗,包括生命科學、材料科學、化學合成、色譜、分析等.
聯絡技術服務