
多孔性配位高分子(PCP)/金属有機構造体(MOF)の基礎
堀毛悟史1, 坂本裕俊3, 杉本雅行1, 福島知宏1, 岸田圭輔4, 梅山大樹1, 北川進1,2
1 京都大学大学院 工学研究科 合成・生物化学専攻
2 京都大学 物質・細胞システム統合拠点(iCeMS)
3 信州大学 エキゾチック・ナノカーボンの創成と応用プロジェクト拠点
4 昭和電工 研究開発本部 研究開発センター
材料科学の基礎 第7号 2012 → PDF版
1.PCP/MOFとは
1.1. PCPを形成する金属イオンと配位子について
1.2. 化学的・熱的安定性
1.3. 多孔体としての位置づけ
1.4. 多孔性構造の特徴
2. PCPの作り方
2.1. PCPをつくるときに考えること
2.2. 従来の方法
2.3. それ以外の(新しい)方法
2.4. ケーススタディ
3. PCPの評価法
3.1. 単離、洗浄方法
3.2. 評価方法 (粉末X線、TG、ガス吸着)
4. PCPの応用・機能
4.1. ガス貯蔵
4.2. ガス分離
4.3. 不均一触媒
1. PCP/MOFとは
固体の構造内部に細孔を持つものを多孔性(ポーラス)化合物と言う。例えば空気中の酸素と窒素を分けるためには活性炭が用いられるが、それは活性炭の構造内部に数 nmの細孔が無数に存在し、その細孔に酸素のみが選択的に取り込まれることを利用している。
このような多孔性材料は有機物や無機物など様々なものが知られているが、この分野において、1990年代後半に新たに見出された材料として、本項で述べる多孔性配位高分子(porous coordination polymer, PCP)がある。PCPは「配位」という名前からもわかるように、金属イオンと有機物の配位結合を利用して多孔性構造を形成する、錯体化学を基盤とする材料である。図1-1に示すように、様々な金属イオンとそれらを連結する架橋性の有機配位子を組み合わせることで、内部に空間(つまり細孔)を持つ結晶性の高分子構造を作る。

図1-1 金属イオンと有機配位子から溶液中でPCPの構造ができるスキーム
これら材料は金属-有機構造体(metal-organicframework,MOF)とも呼ばれ、ガスの貯蔵や分離などの機能を持つ多孔性材料として、この十数年、高い興味を持たれてきた。この化合物群はPCPやMOFの他にもいくつか名称が使われているが、名称に関する議論は関連論文を参考していただきたい1。ここでは以下、PCPという名称を使う。
歴史を見ると、配位高分子(coordinationpolymer)という言葉は1916年にはすでに論文中に登場しており、一世紀にわたって錯体化学の分野では知られていた2。例えば典型的な化合物としてはプルシアンブルーと呼ばれる鉄イオンとシアノ基(-CN基)が互いに配位結合で架橋される三次元骨格がある。この化合物は鉄イオン由来の鮮やかの青色を持ち、染料に昔から使われてきた。また学術的には金属イオン由来の磁気特性が長く研究されてきた。ではなぜこの配位高分子が現在、新たなひとつの分野を作っているのか。その理由は配位高分子が多孔性材料としての機能を示すことが90年代後半に始めて見出されたからである3。配位高分子を構成する金属や配位子の特性に注目した研究は前からなされてきたが、それらが三次元的に組み上がった時、その内部空間に着目し、多孔体としての基礎検討がなされた時から、多孔性材料として急激に注目されるようになった。PCPの面白さは、金属と有機物が示す様々な化学的性質・物理物性のみならず、空間が示す機能にある。この項ではPCPを初めて耳にする研究者、あるいは使ってみたいがとっつきにくいと感じている研究者に対してPCPが持つ基礎的な特性を紹介する。また次の項からは具体的なPCPの合成法、評価法、機能等についてより具体的に説明したい。
1.1. PCPを形成する金属イオンと配位子について
PCPを構成する金属イオンは周期表のほとんどすべての金属で可能であるが、特にCo2+,Ni2+,Cu2+,Zn2+が非常に多い。また架橋性の有機配位子としては酸素ドナー性配位子および窒素ドナー性配位子が多く用いられ、代表的な配位子としては1,4-benzenedicarboxylicacid(14bdc、テレフタル酸、185361)や4,4’-bipyridyl(289426)、imidazole(I202)などが挙げられる。他によく用いられる配位子を図1-2に示している。PCPを合成する際に金属イオンと配位子の組み合わせは1:1である必要はなく、二種類の金属イオンを含むPCP、あるいは複数種の配位子をひとつの結晶構造内部に含むPCPも多数存在する。極端なものだと、結晶構造中に9種類の異なる配位子を同時に入れたPCPも報告されている4。またある金属イオンと有機配位子の組み合わせにおいても、調整法を変えることによって様々な結晶多形ができるため、PCPの取りうる構造は無限にあると言って良い。

図1-2 PCPを構築する金属イオンと有機配位子の例
例えばカルボン酸を配位子として用いる場合、脱プロトン化して-CO2-の形で金属に配位するため、全体として金属カチオンと配位子のみで中性になり、内部に細孔が形成される。一方4,4’-bipyridylのように中性の状態で金属カチオンに配位する配位子は、PCP形成後の電気的中性を保つため、PCPの骨格が正に帯電し、その補償のためアニオンが内部に入る。アニオンの多くは用いた金属塩に由来するものが多く、NO3-やSiF62-など配位性のアニオンを用いた場合は骨格を作る構成素子にもなりうる。
PCPを合成するときに便利な考え方に、SBU(Secondary Building Unit)と呼ばれる構造素子がある。例えば図1-3に示すように、金属イオン一つのみではなく、複数の金属イオンとその回りに配位した配位子の一部を切り取って、金属周りからどの方向に配位結合が伸びうるかをわかりやすくしたものである。有名なものとしては(図1-3a)に示すM2(CO2-)4の組成を持つPaddle Wheel型がある。この場合、カルボン酸が四方向に十字に連結することができるため、例えばテレフタル酸を用いると二次元の層状のグリッド型PCP構造が組み上がる。このようなSBUは数多く存在し、合理的に組み上げた時どのような構造が得られるかの総説も多く発表されている5。多種存在する多孔性材料においても、これほど多彩な設計性を有する物質はなく、PCPの学術的魅力を高めている理由の一つである。

図1-3 SBUの例。(a)Cu2(CO2-)4 paddle wheel型(b)Zn4O(CO2-)6型(c)Fe3O(CO2-)6型。赤:酸素原子、グレー:炭素原子。
1.2. 化学的・熱的安定性
PCPは主に配位結合によって組み上がるため、その結合は一般的に共有結合やイオン結合より弱く水素結合や分子間力より強いとみなすことができる。その構造の形成は図1-1に示すように主に溶液中で自己集合的に行われ、得られた結晶性は非常に高い。また結晶化を工夫することでその多くは単結晶として得られる。そのため単結晶X線解析の手法によって複雑な多孔性構造がこれまでいくつも明らかになっている。一般的にPCPは窒素雰囲気中で300~500℃まで安定であり、この安定性はTGA測定等によって見積もられる。空気中では熱的安定性は下がるが、300℃程度まで安定であるものが多く見られる。
一方で、配位結合によって形成されるため、多くのPCPは水に対して安定性が低い。これは金属-配位子の配位結合が金属-水分子の配位に置き換わってしまうため多孔性構造が崩れることが主原因である。しかしこの安定性もPCPの種類によって大きく変わり、例えば亜鉛イオン(Zn2+)を用いたPCPは水に不安定なものが多いが、アルミニウムイオン(Al3+)やジルコニウムイオン(Zr4+)は非常に耐水性の高い構造を作る。この傾向は古典的な金属イオンの水和の挙動である程度は説明できる。またその一方で、有機配位子として疎水性の高いものを用いたPCPは疎水特性によって細孔内への水の侵入を防ぎ、結果高い耐水性を獲得することもでき、一週間水で還流しても全く崩壊しないものもある。このようにPCPの化学的・熱的安定性はローカルな配位環境と骨格全体の組み上がり方の両方に依存するため、安定性を予測する明確な指針は今のところ乏しい。逆に言えば、様々な金属と配位子の組み合わせと構造の特性をより好適化してゆくことにより、共有結合並の安定な多孔性骨格を作ることも可能である。いくつかの論文では耐水性の評価を系統的に行なっているものがあることを書き加えておく6。またもう一点、PCPは基本的に溶媒に不溶である。多くの有機ポリマーは構造の化学修飾によって溶解性を制御できるが、PCPはその高い結晶構造を維持したまま溶媒に溶けることはない(少なくとも今のところ)。また、耐酸性・耐アルカリ性に関しても配位子の交換が起こるため安定性が低いものが多いが、中には8規定の水酸化ナトリウム水溶液に100℃で浸漬しても崩壊しないものもあり7、耐水性と同様、古典的な錯形成のメカニズムだけでは説明がつかないことが多い。これらの耐水性・耐酸性・耐アルカリ性をより理解することは、PCPを工業的応用に展開するためには必須である。
1.3. 多孔体としての位置づけ
IUPAC(国際純正・応用化学連合)の定義によると、細孔のサイズによって、多孔体は大きく3つの分類ができる。すなわち細孔径が(1)2 nm以下のものをマイクロポア、(2)2 nm~50 nmのものをメソポア、そして(3)50 nm以上のものをマクロポア、と分類する8。これに従うと、PCPは取りうる細孔径が約0.4 nm~6 nmと、マイクロポア~メソポアの領域を持つ。PCPの細孔の形は非常に多彩であり、あるものはボトルネック型の細孔構造をとることによってマイクロポアとメソポアの両方の特性を同時に持つ。またあるPCPは細孔構造に柔軟性を持つものが報告されている。すなわちガス吸着をしていない状態では細孔を持たない、非多孔体であるものが、ガス吸着が進むに従って結晶構造が開き細孔を形成してゆくものである9。このような物質になると上記IUPACの分類は簡単には適用できず、独自の多孔体として検討しなければならない。
ちなみにPCPよりもっと昔から研究されている結晶性の多孔体としては、ゼオライト、メタルホスフェートなどが知られている。ゼオライトはSi4+やAl3+イオンが酸素アニオン(O2-)で架橋され、メタルホスフェートは様々な金属(Al3+,Si4+,Zn2+,Co2+,Ti4+,Zr4+など)と、リン酸アニオン(PO43-やHPO42-)で架橋され、これらの骨格は一般的に無機物とみなされる。一方非結晶性の多孔体あるいは一部結晶性である多孔体としては、活性炭に代表される炭素系材料や、界面活性剤を用いて合成するメソポーラスシリカなどがある。また最近では共有結合を用いた多孔体(Covalent organic framework, COF)など有機骨格のみを利用した(一部)結晶性の多孔性構造の合成も盛んに行われており10、多孔性材料の分類は分野を問わず急速に拡張している。
1.4. 多孔性構造の特徴
上記のように、PCPは既存の多孔体とは異なる構造的特性や物理・化学特性を持ち、今なお急速に発展している。以下では特にいくつかの注目すべきPCPの基礎特性を述べる。
非常に小さい細孔から巨大な表面積まで実現できる
配位子と金属の配位環境周りを変えることで、最も小さなクラスの細孔径(0.4 nm以下)やこれまでの材料で最も高い比表面積(BET表面積~6,000 m2/g)を実現できる。極めて小さな細孔や11、反対に大きな細孔にはいずれの化合物も面白い機能がある。例えば0.4 nmの一次元細孔径を持つPCPである[Cu2(pzdc)2(pyz)](pzdc = 2,3-pyrazine dicarboxylate, pyz = pyrazine)では、図1-4に示すようにアセチレンのような小さなガス分子を細孔中で一列に並べることができる12。これは気相反応触媒などの反応場の機能として興味深い。ガス分子をこのように異方的かつ高密度に内包できることは、ガス分子のサイズとほぼ同等の規則的な細孔があるためである。また、高い空隙率に由来した6,000 m2/gを超える巨大BET表面積PCPにおいては明らかにガス貯蔵において大きな利点を有している。例えばCO2やメタンのガスボンベにおける貯蔵を考えるとき、高い表面積を持つPCP材料を充填させた状態でガスを貯蔵すると、PCPがない場合と比べて10倍以上の貯蔵量を得ることができる。ガスボンベ10本分の貯蔵量を1本でまかなえることになる。これら機能の詳細はあとの項で述べる。

図1-4 [Cu2(pzdc)2(pyz)]がアセチレンガス(C2H2)を規則的に吸着した結晶構造。グレー:炭素原子、ピンク:水素原子
金属由来、配位子由来の物性を付与することができる
PCPの合成において、金属イオンはほぼ何でも用いることができる。最近ではベリリウムイオン(Be2+)13やリチウムイオン(Li+)14、ウラニウムイオン(U4+)15等も金属イオンとして用い、PCP合成の役割を果たしている。骨格にこのような多彩な金属イオンや配位子を利用できることは、骨格に内包できる諸物性の豊かさをもたらす。例えば金属イオンのみを考えると、磁性・伝導性・触媒特性・誘電特性・酸化還元特性・光物性などが機能として考えられる。また配位子の設計としては、金属と配位する部位以外に様々な機能を持たすことができる。不斉点の付与、疎水性/親水性、光応答性など有機化学を範とした物質設計を組み入れることができ、多孔性構造の設計に幅の広がりを与える。
結晶の形状(モルフォロジー)の精密制御ができる
PCPは溶液中において自己集合的に合成することを述べた。合成法の詳細は後に譲るが、溶液中で合成すると、一般的には約数10 μmの微結晶粒子として粉末で得られる。これまで多くの機能発現はこの微結晶粉末を用いて行われてきたが、用途によってはPCPを粉末粒子として扱うのではなく、ナノ結晶化や薄膜化、他の材料と複合化の手法が必要となる。この中で、PCPのナノ結晶の合成法は近年盛んに展開されている。配位結合は金属イオンと配位子の平衡反応であることから、この平衡反応の速度や向きを界面活性剤や補助配位子を用いることによって、結晶成長因子を制御し、結晶のダウンサイジングを行う。例えば例の1つを挙げると、Zn2+と1,4-benzenedicarboxylate(14bdc)と1,4-diazabicyclo[2.2.2]octane(dabco)からなる[Zn2(14bdc)2(dabco)]は図1-5aに示すように四角形の細孔を持つ。このPCPは結晶成長のとき、四方に14bdcとdabcoが伸びてゆきながら連結するが、例えばこの反応時に酢酸を同時に入れておくと、酢酸は金属で架橋されないことから、ある程度結晶成長が起こった時点でPCP結晶外表面を酢酸がブロックし、その結果結晶のサイズを制御することができる(配位モジュレーション法、図1-5b)16。また薄膜化における手法の一つとしては、酸化物などの前駆体の上に直接PCP結晶を成長させる方法がある。
図1-5 (a)通常の[Zn2(14bdc)2(dabco)]の合成および結晶構造(バルク)および(b)合成時に酢酸を用い、結晶成長を抑制したときのスキーム
このように、PCPは格段の設計の自由度を持ち、あくまで基礎学問的な興味から展開されてきた材料である。以下では代表的なPCPの特性、合成法、評価法を具体的に述べながら、研究の進め方を紹介する。
2. PCPの作り方
本項ではPCPをつくるときに考慮するポイント、およびいくつかの合成手法について述べる。前述のとおり、PCPを構築するメインの相互作用は架橋配位子と金属イオン間の配位結合である。したがってPCPの合成は、まず、「適切な構成要素(架橋配位子と金属イオン)を選択」し、そして「それらを配位結合が生じる条件で互いに接触させる」ことが基本である。
2.1. PCPをつくるときに考えること
新規PCPの合成を行う際、組み上がる構造と発現する機能を予想して、架橋配位子と金属イオンの組み合わせを選択する。表2-1の項目を基準にするのが一般的であり、PCPの構造と機能を合理的にデザインできる(図1-2も参照)。
表2-1 PCP設計における金属イオンと架橋配位子の選択基準
また、生成物がどのような形態・粒子サイズで得るかということも考慮する。十分に大きな単結晶(一辺が約0.1 mm以上)が得られればX線回折によってその結晶構造が決定できるので、新規PCPの研究においてはまず、そのようなサイズの単結晶作成に努力が払われる。単結晶は構造解析のほかにも、異方性を示す物性測定などにも用いられることがある。ガス吸着等温線などのバルク物性を調べる際は、試料がそれほど大きな単結晶である必要はなく、調製と取扱いがより簡便な微結晶の粉末試料を用いることが多い。生成した粉末サンプルが目的の結晶構造であるかどうかは粉末X線回折測定により確かめられる。
用いる架橋配位子と金属イオンを決定すれば、目的の形態のPCPが生成するような条件を探索することになる。一般的には、原料試薬を溶媒に溶解させ液相で混合する。ここで考慮する反応条件のパラメータは、金属塩の種類・溶媒の種類・濃度及び濃度比・温度・時間・pH・添加物など様々である。これらのパラメータの違いによって、同じ架橋配位子と金属イオンの組み合わせであっても異なる組成や結晶構造をもつPCPが生成することがある。個々のパラメータが反応系内でPCPの生成に与える影響を考え、反応条件を絞っていく必要がある。同じ種類の配位結合で作られた既知のPCPや、高分子構造ではない錯体(ディスクリート錯体)の合成条件を出発点にして、少数のパラメータを系統的に変化させて反応を行うことが、現実的かつ堅実な進め方の一つであろう。上述のSBUについては、そのディスクリート錯体の合成に関する文献が、骨格構造を構築するという視点でまとめられており、反応条件の選択の参考になる。これ以降は条件に従って試薬を実際に混ぜる。以下では、その「混ぜ方」について述べる。まず、従来から行われている代表的な合成手法である溶液混合法と水熱法について、さらにこれらの効率化や欠点の克服のために開発された、いくつかの新しい手法について紹介する。
2.2. 従来の方法
溶液法
PCP合成で最もシンプルなものは、常温・常圧下で金属イオンと架橋配位子の溶液を混合する方法である。混合する速度を調節することで生成する結晶サイズをコントロールできるので、目的に応じて以下に述べる拡散法や撹拌法を選べばよい。
拡散法では(図2-1a)、内径が7 mm程度のガラスの直管に、金属イオンと架橋配位子の2種類の溶液を層をなすように順番にゆっくりと注入したのちに静置し、液-液界面でゆっくりと溶質を拡散・混合させる。この際の注意点は、重い溶液が下の層になるように先に注入することである。上下で同じ比重の溶媒を用いる場合などは、ただちに拡散してしまい層をなさないことがあるので、下の層になる溶液にあらかじめクロロホルムなどの重い溶媒を少量添加して溶液の比重を高くすると、きれいな液-液界面を形成することができる。金属イオン-架橋配位子間での配位結合の生成が有利(錯体化学の用語で言うと「置換活性」)な条件であれば、液-液界面でこれらが連結して、やがて結晶核が生成する。さらに上下の溶液からの拡散による物質供給速度が適切であれば、生じた結晶核の表面に架橋配位子と金属イオンが交互に配位結合を続けるという結晶成長プロセスを経て、構造解析可能な大きさの単結晶が得られる。この拡散法によって数mmに達する単結晶が得られることもある。単結晶を成長させるのに一般的に数日から数週間静置する。このようにして単結晶が生成する場合は、原料溶液を撹拌しながら混合するとその粉末サンプルが得られる場合が多い。短時間で大量のサンプルが必要とする場合はこちらの撹拌法が簡便である(図2-1b)。

図2-1 代表的なPCP合成法の模式図。(a)溶液拡散法(b)溶液撹拌法(c)水熱合成法
PCPにガス吸着能があることを初めて示した[Co2(4,4'-bpy)3(NO3)4]3aをはじめ、[Cu2(pzdc)2(L)](pzdc = pyradine-2,3-dicarboxylate, L = N donor pillar ligand)で表されるピラードレイヤー構造をもつCPLシリーズ17や、[Zn(R-ipa)(bpy)](ipa = isophthalate, bpy = 4,4’-bipyridyl, R = H, NO2, CH3O…)で表される相互嵌合構造をもつCIDシリーズ18など、多くの単結晶・粉末がこの手法により合成されてきた。これらは架橋配位子の配位部分がピリジル基であり、このような配位子を用いた場合は一般に常温・常圧近くの温和な条件でPCPが生成することが多い。拡散法において、界面に固体が生じているが十分な大きさの単結晶にまで成長しない場合は、混合速度が速すぎるために二溶液接触の初期段階での核生成量が多くなり、ひとつひとつの結晶に原料成分が十分に供給されてないと考えられる。このようなときは溶液濃度を低くしたり、溶液間に溶媒中間層をはさんだりすることで、過剰な核生成を抑え、結晶成長に適した混合速度に調節することで十分な大きさの単結晶を得られることがある。
架橋配位子と金属イオンの溶液を混合しても固体が析出してこない場合は、その条件では配位結合生成の活性化エネルギーを超えられないために反応が進行しない、すなわち置換不活性である可能性がある。このようなときは、溶液濃度を高くしたり、適当な添加剤を加えたり、反応系の温度を上げたり、あるいは後述する水熱合成法を行うなどして配位結合の生成を促進することで結晶化が進行することがある。このような例はイミダゾール系やカルボン酸系の架橋配位子を用いる場合に見られることが多い。アミン系の塩基を加えることによって、配位部位の脱プロトン化が促進され、配位結合に適したカルボキシレート(-CO2-)やアゾレートが系内に生じることにより、結晶の生成が進行することがある19。また、Basolite C300(688614)として販売されている[Cu3(btc)2](btc = 1,3,5-benzenetricarboxylate)は、原料をエタノール中・常温で混合しても生成しないが、これを加熱還流するとその粉末が生成することが確かめられている20。
水熱法
水熱反応(またはソルボサーマル反応)はテフロンの容器に溶媒と原料試薬を入れ、耐圧のステンレスジャケットで密封し、溶媒の沸点以上に加熱して反応を進行させる。(図2-1c)このとき、密閉容器内は溶媒の蒸気圧により高圧となっている。高温・高圧下の溶媒は、溶解性の向上、反応速度の増大、効率的な混合、といった超臨界流体に近い性質を示す。常温・常圧ではどの溶媒に対しても溶解性が低い架橋配位子を用いる場合や、反応速度が低すぎて反応が進行しない金属イオン-配位子の組み合わせの場合に、水熱反応はPCP合成に有効な選択肢の一つとなる。
この水熱反応は、条件によって生成物が構造解析可能な大きさの単結晶にまで成長することも可能であり、これまでに数多くのPCPが合成され、構造決定されてきている。これらの多くは、N,N-dimethylformamide(DMF, 227056 他)またはN,N-diethylformamide(DEF, 186317)溶媒中で調製される。これらは高温で分解してアミン塩基を生成し、これが上述のように配位子の有機官能基の脱プロトン化を促進するために、配位結合の生成が促進されると考えられる。
水熱合成においても、反応条件のパラメータの種類は多く、これらを系統的に変化させて多数の条件を試すことで最適な反応条件を見出すことが必要となる。変化させるパラメータを少数に絞ったとしても、依然として反応試行回数は膨大になりがちである。この反応条件のスクリーニング過程を効率化するために、一連の作業を自動化し同時に複数の条件の反応が行えるようにするスクリーニングシステムが開発されている(図2-2)。このような方法論をコンビナトリアル法と呼び、近年その開発が精力的になされている。原料試薬の分取から、溶液の調製、反応器への注入、反応温度調整、顕微鏡による生成物の外観観察、生成物の取り出し、X線回折による結晶性評価までが機械により自動化されたシステムが開発され、膨大な数の反応条件が極めて短時間で試せるようになり、新規化合物の発見ペースが劇的に上がっている21,22。

図2-2 ハイスループットスクリーニングPCP合成システムの一例。左より天秤、粉末分注用ホッパー、反応用メタルプレート、背面に溶液分注用シリンジが設置されている。
2.3. それ以外の(新しい)方法
近年では、前項で述べた方法に加えて様々なPCPの合成法が開発されてきている。これらにより従来の合成法では得られなかったPCPを合成できることや、これまで難しかった結晶のサイズや形状をコントロールすることやガス吸着や触媒などといった物性を向上させることができることが報告されている。本項ではこれらの新規合成法の中からマイクロ波法、超音波法、固相混合法について簡単に述べる(図2-3)。

図2-3 近年開発されたPCP合成法。(a)マイクロ波法,(b)超音波法, (c)固相混合法
マイクロ波法(図2-3a)
マイクロ波法では、原料となる試薬と溶媒を反応容器に入れ、マイクロ波(300 MHz ~ 30 Hzの電磁波、一般に2.45 GHzのものを用いることが多い)を照射する。安全に使用できる反応装置および容器はいくつかのメーカーから市販されている。マイクロ波は反応容器中の分子を振動させ、その摩擦により熱を発生させる。そのため熱伝導や対流などの影響をあまり受けず、従来の外部から熱を加える方法に比べて系全体を均一かつ短時間で加熱できることが最大の特徴である。これにより短時間でPCPが生成でき、実際に従来の水熱法に比べて約20分の1の時間で合成できることも報告されている。また温度制御により結晶の成長速度の制御が容易であるため、結晶サイズ制御やナノ粒子合成への応用が盛んに行われている。マイクロ波法において特に重要になってくるパラメータは照射時間であり、長時間照射しすぎると過剰にエネルギーを与えることになり結晶性を悪くする原因にもなり得ることに注意が必要となってくる23。
超音波法(図2-3b)
超音波合成法では原料となる試薬と溶媒を反応容器に入れ、超音波(20 kHz ~ 10 MHz程度)を照射する。超音波もPCPの核生成速度に影響を与えるが、マイクロ波法との大きな違いは、超音波は反応容器中の分子を直接振動させるのではなく溶媒と相互作用する点にある。高エネルギーの超音波を照射することで、反応容器中で圧力の変化が繰り返し起こる。この圧力変化により、溶媒が気泡を形成し崩壊するキャビテーションと呼ばれる現象が起きる。その際に約5,000 K、約1,000 barもの高エネルギー場が局所的に形成され、主にこの界面が結晶の核生成の反応場となる。キャビテーションの生じやすさは超音波の周波数や溶媒の種類、温度に大きく依存するため、超音波法を利用する際はこれらのパラメータに注意を払う必要がある。超音波法は金属ナノ粒子の合成によく利用されているように、PCP合成においてもその最大の利点はナノサイズの結晶の合成にある。キャビテーションが起きた場所では核の生成速度が加速される。しかし結晶の成長速度には大きな影響を与えず遅いままである場合が多く、結果としてナノサイズの結晶が得られる。これまでに多くのPCPナノ結晶が超音波法により合成され、報告されており、その中にはガス吸着速度や結晶を構成する分子の運動状態が変わるといったPCPに特有の現象も発見されている24。
固相合成法(図2-3c)
固相合成法は、原料となる試薬を溶媒を用いずに機械的に混合することで合成する手法である。混合には乳鉢やボールミル装置が用いられる。この合成法の利点は、溶媒の無い条件下で合成を行うため溶媒に溶けにくい金属塩や架橋配位子を用いることが出来る点にある。例えば酸化亜鉛(ZnO)などの酸化物は一般に溶媒には溶けにくいためPCPの合成には向いていないが、固相合成法を用いることでこれを金属塩として使用することが出来る。この場合、合成後に生じる副生成物も水のみであるため環境や人体への影響も少ないことも利点として挙げられる25。また、直接試薬を混合するため、短時間(10~60分)で合成できることや反応試薬の混合比を上手く調節することで100%に近い収率が得られ、かつ溶媒を使用しない分だけコストを抑えることもでき、コストパフォーマンスが良いことも特徴である。固相合成法の延長としてLAG(liquid assisted grinding)と呼ばれる合成法がある。LAGは結晶水程度の量の水を加えることで固相反応を促進させる方法で、完全な脱水条件では進行しない反応を促進する効果がある。これは微量に存在する水が固体粒子間および粒子内での反応物質の拡散・混合を促進するためであると考えられている26。
2.4. ケーススタディ
[Cu2(pzdc)2(L)]の合成
PCPの骨格は、2構成要素を適切に選択することによって、その細孔のサイズや表面特性を系統的にデザインできることは上にも述べているが、その例の一つとなるのがここに紹介するcoordination pillared-layer (CPL) structuresである。CPLはCu2+と2,3-pyradinedicarboxylate(pzdc)からなる2次元レイヤーを、直線型のジピリジル架橋配位子(L)がピラー(柱)のように連結することによってできるピラードレイヤー型3次元骨格である。この骨格は組成式[Cu2(pzdc)2(L)]で表され、レイヤー間にはピラー(L)によって区切られた1次元の細孔を形成している。図2-4に示すようにピラー配位子を様々に変化させても共通のピラードレイヤー構造をもつPCPを合成できる。ここではピラー配位子として ピラジン(pyrazine, pyz)を用いてできる([Cu2(pzdc)2(pyz)](CPL-1)の具体的な合成手順を述べる。

図2-4 CPL構造 [Cu2(pzdc)2(L)]の構成要素
まず、2,3-pyradinedicarboxylic acid(H2pzdc)(P56208)は水に不溶なのでナトリウム塩(Na2pzdc)に変換することによって可溶化する。適当量のH2pzdc(16.8 g, 100 mmol)と、それに対して2等量の水酸化ナトリウムNaOH(8.0 g, 200 mmol)を約200 mLの水にゆっくりと撹拌しながら投入する(このとき発熱と強塩基液体になることに注意)。この時点でpzdcは脱プロトン化されている。ここに大量のメタノール(500 mL~)を注ぐと、メタノールに溶けきれないNa2pzdcが白色沈殿として生じる。沈殿がさらに生じなくなるまでメタノールを加えたら、この沈殿をろ別、メタノールで洗浄した後に真空乾燥する。これによりほぼ当量でNa2pzdcが得られる。
つぎにCPL-1の合成反応を行う。三角フラスコに水-メタノールの1:1混合溶媒100 mLを調製し、ここにNa2pzdc(0.21 g, 1.0 mmol)とpyz(ピラジン, P56003)(1.0 g, 12.5 mmol)を溶解させる。また、別に過塩素酸銅(II)六水和物 Cu(ClO4)2・6H2O(215392)(0.37 g,1.0 mmol)を100 mLの水に溶解させたものを調製し、これを滴下ロートに貯め、ゆっくりとNa2pzdc-pyzの配位子溶液に撹拌しながら滴下する(図2-1b)。滴下後、1日ほどで青色沈殿が生じる。これをろ別し、水およびメタノールで洗浄した後に真空乾燥を行う。このときの収量は0.26 gで収率は45%ほどである17。またCPL-1は、拡散混合法による単結晶化(図2-1a)も可能である、上記の配位子溶液およびCu2+水溶液を直管中に層をなすように(Cu水溶液のほうが重いので下に)注入し静置すると1週間ほどで、液界面で青色のブロック状結晶が成長する。CPL-1合成の場合はピラジンを大過剰に用いたほうが結晶性の高いCPL-1粉末が得られる。ピラジン以外のピラー配位子Lを用いる場合は、その量を骨格の組成比当量(Na2pzdcや銅に対して0.5当量)にして上記手順を行えばよい。ただしCPLシリーズの一部のものは単結晶化が困難であり、その結晶構造は基本的に粉末回折パターンのリートベルト解析によって決定されている27。
[Cu3(btc)2](btc = 1,3,5-benzenetricarboxylate)
1,3,5-benzenetricarboxylic acid:H3btc(トリメシン酸, 482749)とpaddle wheel型銅二核クラスターをSBUとする[Cu3(btc)2]の組成で表される化合物は、その原料のシンプルさ、合成の簡便さ、構造の安定性、またCu2+由来の活性な配位不飽和サイトの機能などから、最も多くの研究がなされているPCPの一つであり、前述のようにBasolite C300として市販されている。したがって合成法についても種々研究されており、新しい合成法を開発する際のベンチマーク的化合物であるといえる。
[Cu3(btc)2]は水熱法によって初めて合成されており28、以下にその詳細を述べる。25 mLのステンレスジャケット付きテフロン容器に12 mLのエタノール-水の1:1混合溶媒を調製し、ここにCu(NO3)2・4H2O(229636)5.2 mg(0.022 mmol)とH3btc 2.5 mg(0.012 mmol)を加える。ステンレスジャケットでテフロン容器を密閉し、これをオーブンやオイルバスなどを用いて180℃で12時間加熱する。加熱終了後、十分に容器が冷めてから開けると、青緑色の結晶および粉末が生じている。これをろ別・洗浄して目的の[Cu3(btc)2]が得られる。収率は約60%である。より大きなテフロン容器を用いてスケールアップを行うことは可能であるが、安全のために液体の量をテフロン容器の容積の半分以下にすることに注意しなくてはならない。水熱法のバリエーションとして、[Cu3(btc)2]はマイクロ波によって原料溶液を加熱すると極めて短時間(10分程度)で生成する29。
ここで述べた水熱法のほかにも、単結晶を必要としない場合は、上述の還流法がより簡便である。また工業的には、銅を電極にしてH3btcのメタノール溶液を電気分解することによって迅速に大量合成されている30。
[Zn2(14bdc)2(dabco)](14bdc = 1,4-benzenedicarboxylate, dabco = 1,4-diazabicyclo[2.2.2]octane)
[Zn2(14bdc)2(dabco)]はZn2+と14bdc(テレフタル酸)が形成するpaddle wheel型二核クラスターからなる2次元レイヤーを、dabcoが架橋することで組み上がる3次元骨格を持つPCPである。組成式は[Zn2(14bdc)2(dabco)]で表され、7.5 × 7.5 Å程度の大きさの1次元チャネルを有する。このPCPの最大の特徴はその高い設計性にあり、金属イオンをはじめとしてテレフタル酸誘導体やdabcoとは異なる長さのアミン系配位子を用いることで、基本骨格を変えずに表面特性や細孔サイズを変えることができる。この細孔の特徴を利用したポリマーの細孔内ラジカル重合31、またコアシェル型結晶を合成することでガス吸着特性を制御するといった研究が行われている32。ここではこのシリーズの中で2004年に初めて報告された[Zn2(14bdc)2(dabco)]の水熱合成法について記述する33。
DMF 40 mLをステンレスジャケット付きテフロン容器に入れ、硝酸亜鉛・六水和物Zn(NO3)2・6H2O(228737)1.0 g(3.36 mmol)とテレフタル酸(185361)0.560 g(3.37 mmol)、dabco(D27802)0.187 g(1.67 mmol)を加える。ステンレスジャケットでテフロン容器を密閉し、これをオーブンやオイルバスなどを用いて120℃で2日間加熱する。得られた生成物をろ過し、DMFで洗浄した後、常温で2時間減圧処理することで細孔中にDMFと水を含んだ無色結晶の[Zn2(14bdc)2(dabco)]が80%程度の収率で得られる。ガス吸着測定などを行う際は細孔中のゲスト分子を加熱真空引きにより取り除いてから使用する。[Zn2(14bdc)2(dabco)]は一般に水に弱いため、保存する際はシリカゲル入りのバイアルやデシケータ中で保管することが望まれる。水熱合成法以外では拡散法での合成が行われており、この方法で合成することによりミリメータースケールでの単結晶を得ることができ、結晶の異方性に由来する物性の測定などを容易に行うことができる。
以上、本項においては、PCPを合成するにあたっての考え方、およびいくつかの具体的な合成手法を紹介した。
3.PCPの評価法
本項ではPCPを合成したのち、目的とする化合物が合成できているか、また多孔体として扱う場合の評価法について述べる。評価の過程では洗浄、単離、加熱真空引き、構造決定、ガス吸着測定などのいくつかのステップが必要である。
3.1. 単離、洗浄方法
合成後は、まず生成した固体粉末を単離する必要がある。この単離プロセスの基本的原則は、「原料試薬は溶かすが、生成物を溶かさない」溶媒で洗浄し、固液分離・乾燥を行うことである。また、単離する場合にはその合成反応を行った状態が重要である。大きく分けてPCPの合成においては、1)有機溶媒、イオン液体などに溶けた構成要素が溶液中から自己集合することにより得る溶液合成法、2)固相中で金属イオンと配位子を混合し得る固相合成法があり、これらそれぞれにおいて合成直後の生成物の状態が異なる。
1)の溶液合成法の場合には、粉末結晶であるPCPは合成後、溶媒と共存状態にある。PCPの固体粉末を得るためには、溶媒と固体を取り除く必要がある。仮に溶媒を全て加熱真空引きにより取り除くと、未反応の金属塩や有機配位子が含まれてしまい目的とする生成物が不純物を含んだ状態で得られてしまう。そのためこれら溶媒を加熱真空引き以外の処理により取り除く必要がある。基本的に多くのPCPは粒子径が数マイクロメートル程度と大きいサイズで得られるため、粉末結晶をロートでのろ過により単離することができる。ただし合成時に粘性の高い溶媒を用いた場合には単純なろ過をしても、粉末結晶の周りに溶媒が存在し、単離ができない。このような場合には合成溶媒との親和性の高い低沸点の溶媒を用いて洗浄することで粉末結晶を回収することができる。例えば親水性の溶媒の場合にはメタノールを、疎水性の溶媒の場合にはヘキサンあるいはベンゼンを用いる。また粒子サイズがろ紙を通過してしまう程度に非常に小さな場合には、ろ過による単離は困難であるため、遠心分離法を用いる。粒子を含む分散溶媒を高速回転させることにより沈降させ、上澄みの液体成分を取り除くことが可能となる。2)の固相合成法は反応系内に必ず未反応の反応試薬を含んでいるため、上に述べた洗浄操作を必要とする。
3.2. 評価方法 (粉末X線、TG、ガス吸着)
単離を行った後に構造の同定、細孔特性評価を行う。粉末X線測定からは得られた粉末結晶が単結晶と同様の構造を有するかを確認し、TGからは得られた化合物の熱的安定性および合成直後のものにおいては細孔内にどの程度の量の合成溶媒が入っているかの確認、そして加熱処理温度の推定が行える。ガス吸着測定からは生成物の持つ細孔特性、特に細孔表面積(BET表面積、Langmuir表面積)の同定、細孔サイズの分布が主に分かる。
PCPの結晶構造評価
PCPは基本的に結晶性固体であるため、その同定においてX線構造解析は重要である。PCPの多くは単結晶として調整することができ(図3-1)、X線による厳密な構造解析が可能である。近年の測定技術の向上により、小さな結晶であっても構造解析が可能となってきているおり、0.05~0.4 mm角程度の大きさを持つ単結晶であれば十分に測定・解析を行うことが可能である。測定の際には単結晶をガラス棒などの非晶質の先端に固定し、X線に露光をすることで測定を行う。PCP特有の測定としては例えば、溶媒雰囲気下での測定が挙げられる。一部のPCPは空気雰囲気に曝露することにより空気中の水分と反応し、単結晶性が失われることがある34。また酸化されやすい配位中心を持つ金属種(Fe2+、Cr2+、Cu+)が錯体中に含まれている場合には空気中の酸素と反応してしまう場合がある。このような場合はガラスキャピラリーと呼ばれる先端が閉じられた中空のガラス管を用いる。ガラス管内部を溶媒雰囲気で満たし、結晶を内部に閉じ込めた後に余分の溶媒を取り除くことで溶媒雰囲気にすることが可能となる。このガラスキャピラリーをバーナーにより封じきることで単結晶性を失うことなく測定が可能となる。

図3-1 PCPの単結晶の写真(例)
また基本的に結晶構造解析は-100~-50℃程度の低温条件で測定することにより結晶中の分子運動を抑制し、原子は位置を正確に決める必要がある。ただ一部のサンプルでは室温以上の温度において測定を行う場合がある。たとえば、PCPが熱的に相転移を示す場合には温度を変えて測定を行うことでそれぞれの構造状態を明らかにすることが可能となる35。また溶媒分子を脱離することにより錯体自体が構造変化を示す場合がある33。ガラス管の先端に結晶を接着剤により固定し、窒素吹きつけにより加熱処理を行うことで細孔内部にある溶媒を除去することが可能となる。
測定の後に得られる解析に必要なデータの含まれたファイルを、Crystal Clear、ydkrなどの結晶構造解析ソフトウェアを用い、結晶構造のモデルを実測の電子密度情報、実際に使った金属イオン・配位子情報と照らし合わせながら形作ってゆくことにより、単結晶構造が得られる。なおydkrはフリーウェアであり、現在のところ日本語のみ対応ではあるが、ユーザーインターフェースも使用しやすくお勧めである。
結晶性粉末の評価
結晶粉末を得た場合には初めに粉末X線回折測定を行う。粉末X線回折パターンはその名の通り、粉末サンプルにX線を照射した時、結晶格子によって回折されたX線の強度の、観測角度に対する依存性を表したものである。格子長と同程度あるいはそれ以下の長さであるCuKα(波長:1.541 Å)などの特性を用い、Braggの式:λ = 2dsinθ(λ:X線の波長、d:結晶格子から定義される面間隔、θ:回折角(2θ)の半分)に基づいて回折ピークが観測される。この粉末X線回折パターンは、そのサンプルの結晶構造を直接反映した情報を与えるが、これからただちに結晶構造を決定できるわけではない。実際には得られたパターンが正しいかどうかは単結晶X線構造解析により得られた構造ファイル(CIFファイル)と一致するかどうかから判断をすることが多い(図3-2)。たとえばMercuryなどのソフトウェアを用いることでシミュレーションをすることができる。サンプルホルダーに粉末サンプルを乗せ、θ-2θ方式で測定を行う場合にはサンプル量に注意を払う必要がある。サンプル量が多過ぎる場合や少なすぎる場合、また、粉末結晶の盛り方が均一(等方的)でない場合、シミュレーションと実験結果が一致しなくなってしまうことがある。
例として、図1-5にも示した[Zn2(14bdc)2(dabco)]を挙げる。[Zn2(14bdc)2(dabco)]は溶媒中での反応により粉末結晶として得られる。このPCPは空気中の水分に対して弱いので、ろ過による単離では錯体自身が壊れてしまうことがある。そこで遠心分離により、粉末結晶と溶媒を分離しCHCl3により洗浄操作を行う。図3-2に粉末X線パターンを示すが、単結晶の結晶構造ファイルから得られるシミュレーションパターンとよい一致を示しており、得られた粉末試料がちゃんと合成・単離ができていることがわかる。

図3-2 [Zn2(14bdc)2(dabco)]における(a)実測の粉末X線パターンおよび(b)単結晶から得られる粉末X線パターンシミュレーション
また例は少なくなるが、単結晶構造情報がこれまで得られておらず、粉末X線測定の結果から構造解析を行うことがある。これを行う場合には得られた粉末X線のピーク位置からセルパラメータを推定し、原子配置からピーク強度を再現できるようにシミュレーションを行う(Rietveld解析)36。Rietveld解析にもとづく構造の場合、元素分析などの結果と照らし合わせて構造、組成の推測を行うことが重要になってくる。これらに関しては「粉末X線回折の実際(第二版)」(朝倉書店)という本が詳しい。
またマイクロメートルサイズの粉末結晶のみならず、PCPをナノ粒子化した場合や、薄膜化した場合にも粉末X線は威力を発揮する。一般に結晶のサイズが小さくなると、回折ピークの半値幅は広くなることがシェラーの式より知られている。そのため半値幅を見積もることにより、電子顕微鏡測定などを行う前にナノ粒子化できているかどうかを簡便に予想することができる。また近年ではPCPが薄膜化できることが知られている37。PCPの薄膜化を行う上で、全て単結晶的に薄膜化されているか、それとも結晶が多く集まって2次粒子的に薄膜化されているかを確認する。これらはその薄膜の強度や気体透過能などにおいて重要である。異方的に結晶が配向している場合、配向した指数面に由来するピークのみが出現し他のピークは消滅する。ここから薄膜の配向を決定することが可能となる
細孔内部に取り込まれた分子の挙動は熱重量分析(TGA)を用い評価することができる。TGAはアルゴンや窒素などの反応不活性なガスを吹き付けながら、一定速度での熱上昇に伴う重量減少を測定する簡便な手法であり、多くの場合熱のやりとりを測定する示唆熱分析(DTA)も付属している。図3-4に示すように、TGAからPCPにどの程度の数の溶媒分子が含まれているか、溶媒分子の除去にどの程度の温度が必要かということがわかる。DTAからはゲスト分子が熱的に放出される場合は吸熱ピークが、PCPの構造が崩壊する場合は発熱ピークが観測される。TGAとDTAを詳しく見積もることでたとえば溶媒分子の脱離や相転移が起きているかどうかの確認などが可能となる。またTGAからゲスト分子がどの程度強く拘束されているかを見積もることができる38。たとえば、ゲスト分子との親和性が非常に高い場合には掃引速度を速くすることで溶媒脱着の温度が高温側にシフトするのに対し、ゲスト分子との親和性が低い場合には溶媒脱着の温度があまり変化しないことが知られている。
図3-3に[Zn2(14bdc)2(dabco)]のTGカーブを示す。図3-3aに示されるように細孔内部にCHCl3を含んでいる場合には、35重量%程度の重量減少を示す。これを加熱真空引き処理により、溶媒を取り除くと、図3-3bに示すように錯体は280℃程度まで重量減少を示さず、溶媒分子を取り込んでいないことまた熱的に安定であることがわかる。
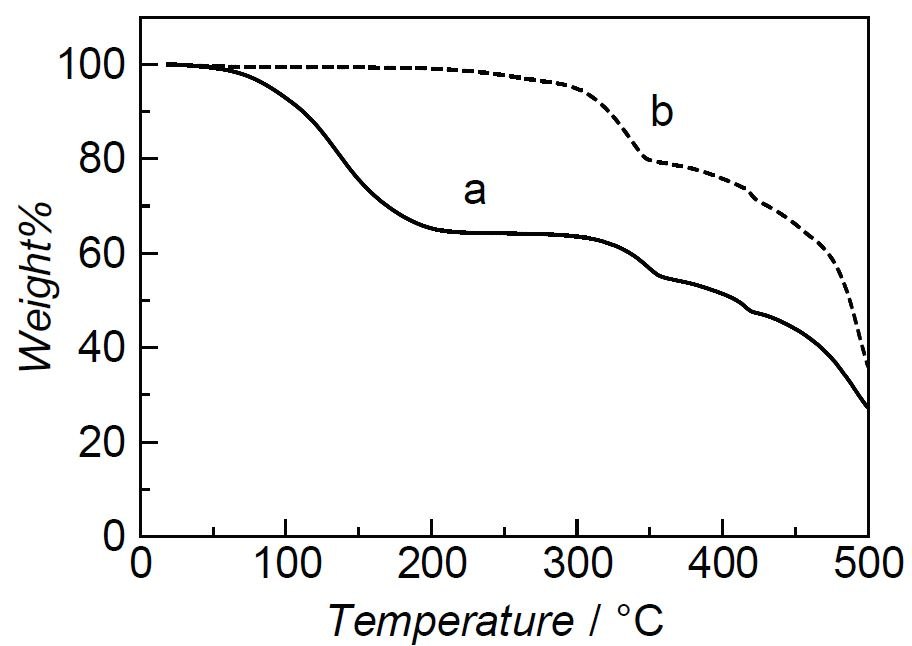
図3-3 [Zn2(14bdc)2(dabco)]の(a)合成直後および(b)デガス処理のサンプルのTGカーブ。(a)における100~160℃付近の重量減少は細孔中の合成溶媒による。
粒子径の評価
結晶の粒子径の制御は固体表面積の増加をもたらすため、触媒能の向上やガス吸着速度の増加をもたらし、粒子径の評価をおこなうことで機能評価ができる面も大きい。また近年ではPCPとナノ粒子を複合化させる例が多く報告されており、これらを顕微鏡観察により同定を行う場合がある。粉末固体である粒子径の評価は顕微鏡を用いて行うが、通常の光学顕微鏡では光の波長程度の大きさの粒子になってしまうと、原理上測定ができなくなってしまう。そのため電子顕微鏡を用いる。電子顕微鏡とは電子線を使った顕微鏡であり、測定は真空下で行う。そのため測定前には加熱真空引きを行っておくことが必要になる。走査型電子顕微鏡(SEM)は電子線の加速電圧を低くすることで表面近傍の観察を行う手法であり、透過型電子線顕微鏡(TEM)は加速電圧を大きくすることで、電子線を透過させて結晶の内部構造を探る事ができ、近年では細孔構造を直接観察することも可能になっている11。ただPCPは金属イオンと有機配位子からなるため、TEM測定において電子線のエネルギーが高すぎる場合、結晶が壊れてしまうことがある。
またナノ粒子と複合化した場合には金属種の分散状態を把握することにより、より高い機能設計が可能となる。存在核種の分析も可能なエネルギー分散型X線検出器(EDX)を利用することで、SEM-EDX、TEM-EDXなどの測定から、細孔内部に存在するか、表面に存在するかなどの評価が可能である39。
前処理方法
ガス吸着測定前には細孔内部に取り込まれた分子を取り除く前処理が必要である。前処理は一般的にTG測定より見積もられた温度で加熱減圧処理を行うことで細孔内部に取り込まれた溶媒分子を取り除くことができる。ただし、ときによっては前処理時に生じる毛管張力現象により多孔性構造が壊れることや、溶媒分子が細孔内部に取り残されて後の吸着測定で想定される値よりも低い表面積を示すことがある。これは細孔内部に凝集していたゲスト分子が気体になって出て行く際に、表面に吸着した液相分子に由来する毛管張力がはたらき、この圧力がしばしば多孔性構造を破壊する。このような場合には超臨界二酸化炭素を用いた活性化手法が有効である。超臨界二酸化炭素でゲスト分子を置換し、そののち超臨界二酸化炭素は常温で減圧することにより超臨界-気相状態へと変化するため、結果として高いガス吸着量を有するPCPが得られる。
前処理後に多孔性構造が壊れている場合もあるので、粉末X線、TGAを測定し確認する。またこれらの処理のみでは溶媒分子が抜けていないこともしばしばある。前処理後に粉末固体をIR/ラマン分光により調べること、あるいは錯体を強酸・塩基により崩壊させた後に溶液NMRを測定することにより溶媒分子の存在を確認することができる。また溶媒脱着により構造が変化する場合もある。このような場合は、再度溶媒曝露することで、溶媒を吸着させ、結晶構造が元に戻るかを確認する。
ガス吸着測定
PCPの細孔特性を知ることにより吸蔵、分離、触媒や輸送などといった様々な機能発現が可能となる。PCPの細孔特性を知るための基礎的な測定としてガス吸着測定が挙げられる。吸着測定から知り得ることとしては細孔内部の表面積、細孔の大きさの分布、細孔へのゲスト分子の親和性、分子選択制などが挙げられる。吸着現象は気体状態から細孔内部へと分子が吸着(相転移)する現象である。多くの物理吸着は発熱的であるため、結果低温では分子は吸着されるが高温では分子が脱着される。またバルクでの気体から液体あるいは固体へと相転移する場合と比較して細孔内部ではホスト-ゲスト相互作用に由来し、低圧力で凝集できる。これはマイクロ孔の場合には、これらの固体表面からのポテンシャルが重なり、深い引力ポテンシャルを形成する。そのため分子が引き寄せられて吸着する。このポテンシャルは細孔径が小さくなるほど、深くなる。後述するようにこれを利用して細孔径分布などを算出することができる。
吸着等温線は、一定温度における圧力と吸着量の2次元プロットである。吸着量は様々な単位で表現されるので、異なる単位の吸着等温線間で比較する場合は注意しなくてはならない。一般に前処理後のサンプル重量を単位として、吸着された物質の量としてml (STP)/g(標準状態298 K, 1 atmにおける気体の体積に換算した単位)、mmol/g、mg/g、など、実用的には、吸着ホストに対する吸着量の重量パーセント(重量%と表される)が使われる。また、とくに結晶構造の決まっているものに対しては単位格子あたりの分子数(molecules/unit cell)を計算することができ、ホスト-ゲスト間の相互作用の規則性の有無を推定することができる。また横軸の圧力は、絶対圧のほかにも、飽和蒸気圧に対する相対圧や、熱力学的な解析で要請される化学ポテンシャルでの表記などがある。吸着等温線の測定では、液体窒素(77 K)や液体アルゴン(87 K)、ドライアイスとメタノールのシャーベット(195 K)を用いた恒温槽にサンプルを浸しながら温度一定の条件下で測定を行うことが一般的であるが、近年は吸着測定装置に特化したクライオスタットによる温度コントローラーが開発されており、幅広い温度条件での測定が可能となっている。吸着測定の測定原理は、定容法、重量法という大きく分けて二種類の手法があり、いずれの手法においても、サンプルを測定セル内に密閉し、バルブ操作によりサンプルセル内のガス濃度(圧力)を変化させる。サンプルはセルに満たされたガスを吸着するにつれて、セル内のガス濃度(圧力)が低下していく、とともにサンプル重量が吸着量分だけ増加する。このセル内圧力およびサンプル重量の変化は、時間につれてゆるやかになり、やがて変化しなくなる。この状態を「吸着平衡状態」と呼ぶ。圧力操作時点からこの吸着平衡状態までが吸着等温線で1点プロットするためのプロセスであり、定容法では圧力変化量から気体の状態方程式を用いることによって吸着量を求め、重量法ではその重量変化がそのままこのステップでの吸着量となる。この量は1ステップでの吸着量である、これを繰り返し、足し合わせることによって、吸着等温線は「累計」吸着量として表される。したがって、各ステップ単位では誤差が少なくても、累計すると大きな誤差になりかねないので、各ステップで正確な吸着量を求めることが求められる。脱着等温線は、圧力変化のプロセスが吸着等温線と逆になる。Bel-Japan、Quantachrome、Micrometricsなどの会社から自動測定装置が販売されている。
吸着測定の際には吸着管の重量を量った後に、試料を吸着管に入れ、加熱真空引き処理したのちに重量測定を行うことで、正味のサンプル重量を算出する。[Zn2(14bdc)2(dabco)]の場合にはサンプルを入れた後に120℃にて加熱真空引き処理を12時間程度行う。その後にサンプルを液体窒素温度に冷却し、適切に窒素ガスを導入し、圧力変化を追うことにより吸着測定を行う。図3-4に[Zn2(14bdc)2(dabco)]の窒素吸着等温線を示す。このサンプルは、10-4~10-3の相耐圧領域にて、吸着を示すことがわかる。この領域において飽和吸着量に達しており、吸着量は380 mL (STP)/g程度であることがわかる。

図3-4 [Zn2(14bdc)2(dabco)]の77 Kにおける窒素(N2)ガス吸着等温線。●:吸着、○:脱着
窒素吸着はもっとも標準的な測定である。これは液体窒素が手に入りやすいことと、窒素分子が比較的単純な構造をしているために双極子相互作用などの強い相互作用を持ちにくいためである。窒素吸着の結果から細孔径分布を求めることが可能であり、1 nm以下でのミクロ孔では相対圧(P/P0)を10-5から10-2の範囲、メソ孔の場合はP/Psが0.5~0.9付近をもとに細孔径を見積もることができる。ただし細孔径は十分大きいにも関わらず低温では窒素を吸着しないPCPも報告されており40、新規化合物でガス吸着能を示すかどうかを確認するためには、二酸化炭素吸着なども必要となってくる場合が多い。このような新規化合物探索のもと吸着量の多い錯体が見つかっている。例えば[Mg2(dobdc)](dobdc = 2,5-dioxido-1,4-benzenedicarboxylate)は25℃、0.1気圧の状態で19.8重量%の二酸化炭素吸着量を示す41。また[Zn4O(bbc)](bbc = 4,4’,4’’-(benzene-1,3,5-triyl-tris(benzene-4,1-diyl))tribenzoate)は常温50気圧のもと70.6重量%の二酸化炭素を吸着する42。また細孔内部に取り込まれた分子がどの程度強いのかはクラウジウス-クラペイロン式により、見積もることが可能である。一部の錯体では二酸化炭素への吸着熱が90 kJmol-1を超えるものも見つかっている43。また一方でIUPACによる吸着等温線8には分類することができない等温線も見つかっている44。このような場合には従来の条件とは異なる測定条件を設定しなければならない。
他の細孔特性の評価
PCPは細孔内部に配位不飽和な金属サイトに由来する酸点や、アミノ基などの塩基点を含む場合がある。このような構造は触媒能を示す場合があり、酸点塩基点の強さを測定することが重要となる。そのような場合にはプローブ分子を細孔内部に導入し、それらの結合状態の違いにより推定することが可能となる。酸点を評価するためのプローブ分子としてはアセトン、ピリジン、NH3、COなどが上げられる。例えばアセトンが酸と相互作用した場合カルボニル部位の酸素原子と相互作用して、二重結合性の低下が確認される。その場合IR測定によりはカルボニル伸縮の低波数シフトを示す。また固体NMRが使用できる環境にあるのなら非常に強力な手段となり得る。核種のまわりの電子分布が酸塩基としての特性を強く反映するので、NMRによる化学シフトによる評価は強力な手法であるといえる。13C、15Nなどでラベルし、特定の各種の化学シフトを見積もることにより、実際酸塩基特性がどの程度異なるかを考えることができる。また分子運動性の評価は固体NMRで多くの知見が得られる手法である。
以上本項においてはPCPの評価方法を中心に述べた。サンプルの回収、前処理に始まり、ガス吸着測定からプローブ分子を用いた手法について述べた。PCPの評価は後の機能発現に重要な役割を果たすことを忘れてはいけない。
4. PCPの応用・機能
4.1. ガス貯蔵
金属イオン及び有機配位子の選択によって、従来の多孔性材料よりも細孔表面積が大きく、空隙率の高い材料を合成できることから、PCPを用いたガス貯蔵は応用開発のメインストリームと言える。様々なガスの貯蔵が検討されており、例えば最も水素を貯蔵する化合物としては、Zn2+と4,4’,4’’-(benzene-1,3,5-triyl-tris(benzene-4,1-diyl))tribenzoate(元となる酸, 686859)および4,4’-biphenyldicarboxylate(元となる酸, 225266)からなるMOF-210が17.6重量%(77 K、80 bar)の水素を吸蔵するという報告がある42。しかしながら、水素においては実用化目標との乖離がまだまだ激しく、水素貯蔵に関する研究開発は基礎段階にとどまっている。現在は、水素貯蔵検討で得た知見を生かし、より実用化への可能性が高い、メタン貯蔵や二酸化炭素貯蔵に開発の目標が移っている。メタン貯蔵の一例を挙げると、[Cu2(H2O)2(adip)](adip = 5,5’-(9,10-anthracenediyl)di-isophthalate, PCN14)45において、220 cm3(STP)/cm-3(290K, 35 bar)というメタン貯蔵量を記録しており、これはアメリカ合衆国エネルギー省(United States Department of Energy)が設定した目標値(298K, 35 barにおいて 180 cm3(STP) cm-3)を見かけ上クリアしている。実際に利用する際は、粉体もしくはペレットの形で使うことが想定され、その際には、PCP粒子間に死容積が存在するが、この論文ではその点については考慮されていないため、見かけ上と表現している。メタン貯蔵については、その吸着原理にまだ不明な点があるため、吸着量を上げる検討とあわせ、今後も盛んに研究開発が行われていくものと予想される。
4.2. ガス分離
貯蔵と同様、多孔性材料であるPCPは分離材としての応用を目指した研究開発も盛んに行われている。バイオガスの精製や二酸化炭素の排出抑制を目的とし、CO2の分離に関しては特に報告例が多い。各種金属塩と2,5-dioxido-1,4-benzenedicarboxylate(dobdc)(元となる酸, 382132)から合成される[M2(dobdc)](M = Mg2+, Mn2+, Co2+, Ni2+, Fe2+, Zn2+, MOF-74)は、約1 nmの1次元細孔を持ち、細孔表面に上記金属イオンの配位不飽和サイトが露出している。中でもMg2+から合成される[Mg2(dobdc)]を充填した吸着管に、体積比CO2:CH4=20:80の混合ガスを流し、出口のガス組成を測定すると、CO2が除去され、高純度のCH4が得られる46。吸着量も従来材料に比べて多い一方、CO2の吸着エネルギーはそれほど高くないため(39 kJ/mol)、比較的マイルドな条件で再生、再利用可能な事が特徴である。また、このPCPはCO2のほかにも、SO2、NH3、ethylene oxideなどの分離除去についても報告されている。
再生エネルギーの低減化については、ガス分子との相互作用の設計以外にも、PCPのゲート型の吸着挙動に着目した分離材の開発も行われている。Cu2+、4,4’-bipyridyl(bpy, 289426)および4,4’-dihydroxybiphenyl-3-carboxylic acid(H3dhbpc)からなる[Cu(dhbpc)2(bpy)](図4-1)は、CO2を段階的に吸着し、まず34 mLg-1吸着したのち、加圧することでさらに80 mLg-1吸着する47。脱着に着目すると、大気圧(0.1 MPa)以上の圧力において2段階目で吸着したCO2を脱着する。一方、メタンは1 MPaにおいても全く吸着しない。この材料を圧力スイング吸着法(PSA法)の吸着材として利用することで、常圧にするだけで吸着材を再生することができ、従来再生に必要であった真空ポンプを用いる必要がなく、ランニングコストの省エネ化につながると期待されている。
PCPを含む多孔性材料において、混合ガスからの分離能を調べるためには、貯蔵より評価法が複雑になる。分離特性の評価においては、単成分ガスの吸着等温線をそれぞれ比較するだけでは不十分であり、混合ガスを用いた平衡状態の共吸着状態(吸着量等)、および混合ガスを流通させてどれだけ分離吸着できるかを時間とともに追う測定(破過曲線測定)を行わなければいけない。

図4-1 [Cu(dhbpc)2(bpy)]の合成スキームおよび(a)結晶構造(b)273 KにおけるCO2およびCH4のガス吸着等温線(●:吸着、○:脱着)
分離対象としては、CO2/CH4以外にも、炭化水素の分離等への応用も期待されている。一例として、エチレン精製について述べる48。Zn2+とbenzimidazolate(-zole, 194123)から合成されるゼオライト様の構造をもつ[Zn(benzimidazole)2](ZIF-7)と呼ばれるPCPはゲート型の吸着挙動を示す錯体であるが、プロピレンよりもプロパン、エチレンよりもエタンをより低圧から吸着する。これは従来の多孔性材料では見られない特徴であり、この特徴を利用し、エチレン/エタンの混合ガスにおいて、エチレンではなく、エタンを選択的に吸着し、エチレンの純度を上げることができる。このパラフィン選択性は、プロピレン/プロパンにおいても同様である。
また、上記以外にも酸素富化を目指したO2/N2分離についても報告例がある。特に、従来材料では困難なO2選択吸着について、Cr2+と1,3,5-benzenetricarboxylate(btc)(元となる酸=トリメシン酸, 482749)からなる[Cr3(btc)2]において高い選択性が発現している(図4-2)49。この錯体は、Cr2+が配位不飽和な状態で細孔中に露出しており、酸素原子と電荷移動を伴う相互作用をするため、高いO2選択性を発現している。また、酸素原子は50℃で真空引きすることで取り除くことが出来、吸着材として再利用可能であることが報告されている。
このようにいくつかのPCPは既存の材料には見られない機構に基づくガス分離挙動が見られ、既存材料の代替としてのみではなく、これまで困難とされてきたガス分離の分野の開拓が期待される。

図4-2 [Cr3(btc)2]の合成スキームおよび不飽和Cr2+サイトにおける酸素ガスの吸着(室温)。酸素ガス(O2)は赤色の球で表している。
4.3. 不均一触媒
貯蔵剤、分離剤としての利用が実用化を見据えた研究が進む中、近年では触媒としての利用も精力的に検討されている。大きな表面積の活用として、その表面における触媒反応は古くから志向されてきた。ゼオライトなどの固体触媒に比べ、比較的マイルドな条件で合成されるPCP触媒は、安定性では劣る一方、極めて高度な設計性を持っているのが特徴である。有効な反応空間を残したまま配位子のサイズや官能基を系統的に変更したり、不斉部位を高度に集積・配列させたりすることは他の材料ではなかなか難しいため、PCPはオーダーメイド可能で付加価値の高い触媒として位置づけられるだろう。この触媒作用は、配位子か配位不飽和な金属サイトが活性点となる場合がほとんどである。多孔体であるPCPは内表面に対する外表面の割合は非常に小さく、反応活性点のほぼすべては細孔内部に存在する。よって反応基質は細孔内に拡散して行き、そこで反応することになる。細孔サイズよりも大きい基質は活性点にアクセス出来ず、それより小さい基質だけが反応するため、基本的にPCP触媒にはサイズ選択性が備わっている。
PCPの骨格が作り出す効果は単純にアクセスできる反応基質のサイズを制限するだけではない。例えば、図4-3に示す触媒作用をもつメタロリガンドを組み込んだ触媒、[Zn2(bpdc)2L](bpdc=biphenyldicarboxylate, L=(R,R)-(-)-1,2-cyclohexanediamino-N,N’-bis(3-tert-butyl-5-(4-pyridyl)salicylidene)MnIIICl)では、そのメタロリガンド単独の触媒活性を上回る性能が報告されている50。この効果は「confinement effect(基質束縛効果)」とよばれ、多数の活性点が集積することによる相乗効果と考えられている。

図4-3 [Zn2(bpdc)2L](bpdc=biphenyldicarboxylate, L=(R,R)-(-)-1,2-cyclohexanediamino-N,N’-bis(3-tert-butyl-5-(4-pyridyl)salicylidene)MnIIICl)の(a)結晶構造および(b)2,2-dimethyl-2H-chromeneの不斉エポキシ化反応。
触媒の機能のうち、特に注目されているのは不斉選択性である。一般に固体触媒による不斉選択性が均一系錯体触媒に比べ劣るのは、活性点近傍の不均一性が大きく精密な立体制御ができないためである。ところが結晶性多孔体であるPCPは、原子配置の均一さは通常の錯体触媒と全く変わらない。従って原理的には、固体でありながら均一系の錯体触媒と同等か、それ以上の不斉選択性が見込まれる。実際、非常に高い不斉選択性をもつ触媒特性がすでにいくつか報告されている51。不斉反応には白金などのレアメタルが使われることも多いため、多孔性構造にうまく組み込むことで不均一触媒として利用できるようになれば、新しいタイプの固体触媒として注目されるであろう。
また触媒としての利用するアプローチには、フレームワーク自体に活性を持たせる以外にも、触媒の担体として用いるという方法もある。これまでに、金ナノ粒子やポリオキソメタレート、ポルフィリンや有機触媒などを担持・分散させた複合体が検討され、触媒活性が報告された。PCPは細孔のサイズや化学的性質を変えることで様々な物質を担持できるため、よく知られている触媒を規則的な空間に配置したときの効果なども興味深い側面である。
5. まとめと展望
これまでに述べてきたように、PCPは周期表を自在に横断しながら様々な金属イオンと有機配位子で多孔性構造を作りうる。数年前までは初めに述べたようにZn2+やCu2+など限られた金属イオンが研究対象であったが、現在は非常に多彩となっており、例えば酸化還元能を示すFe2+やCr2+を用いた多孔性構造も報告され、金属イオン由来の高い活性を持つ細孔を形成する。配位結合を用いた多孔体という点では、以前から研究されているメタルホスフェートもPCPと言うこともでき、多孔性材料は無機・有機問わずボーダーレスになっている。研究の目的によって柔軟に多孔性材料の種類を選ぶことが重要になっている。
PCPを初めて扱うとき、既知の化合物を論文に記載されている手順に従って合成と解析を行う場合が多いと考えられるが、PCPは一見非常に合成が簡単に見えてなかなかうまくできないことがある。また評価においても順を追って必要十分な測定を行い解釈しなければ、最大のパフォーマンスは引き出せない。例えば粉末X線で以前のレポートと一致していても、X線は結晶性のところしか回折を示さないため、含まれているかもしれない不純物は分からない。合成時の洗浄やTG測定、元素分析測定等を適切に併用して初めて高純度・高結晶性のPCPを得ることができる。洗浄が不十分な場合、細孔表面積が半分以下になってしまうこともあるため、十分な注意が必要である。
またPCPの解析においてはX線測定のための単結晶の作成が極めて重要であるが、この単結晶作成においても、各研究グループで様々なノウハウがある。PCPに限ったことではないが、単結晶の生成は反応溶液の濃度や温度はもちろんのこと、例えば加熱中の熱対流や結晶核のコントロールが鍵をにぎる場合もあり、実験システムを変えるとできるはずのものができないことは多々ある。ここで述べたことがスムーズな実験の一助になれば幸いである。
PCPが多孔性材料として見出されてすでに15年ほど経っているが、いまだに新たな結晶構造の論文報告は増える一方である。また工業的応用を見据えた研究は、ここ数年以内に盛んに見られるようになっており、使用する際の安定性の改善や知見の蓄えはますます重要になる。ガス貯蔵やガス分離は実用化に近い分野と言えるが、多孔性材料の最近の多様性を考えると、既存の多孔性材料が担っている機能の改良だけではなく、既存材料で実現できない新たな機能分野をPCPで開拓していくことがより一層望まれる。
引用文献
- Batten, S. R.; Champness, N. R.; Chen, X. M.; Garcia-Martinez, J.; Kitagawa, S.; Ohrstrom, L.; O'Keeffe, M.; Suh, M. P.; Reedijk, J. Crystengcomm 2012, 14, 3001.
- Shibata, Y. Journal of the College of Science, Imperial University of Tokyo 1916, 37, 1.
- (a) Kondo, M.; Yoshitomi, T.; Seki, K.; Matsuzaka, H.; Kitagawa, S. Angew. Chem. Int. Ed. 1997, 36, 1725;(b) Li, H.; Eddaoudi, M.; Groy, T. L.; Yaghi, O. M. J. Am. Chem. Soc. 1998, 120, 8571.
- Deng, H.; Doonan, C. J.; Furukawa, H.; Ferreira, R. B.; Towne, J.; Knobler, C. B.; Wang, B.; Yaghi, O. M. Science 2010, 327, 846.
- Eddaoudi, M.; Moler, D. B.; Li, H. L.; Chen, B. L.; Reineke, T. M.; O'Keeffe, M.; Yaghi, O. M. Acc. Chem. Res. 2001, 34, 319.
- Cychosz, K. A.; Matzger, A. J. Langmuir 2010, 26, 17198.
- Park, K. S.; Ni, Z.; Cote, A. P.; Choi, J. Y.; Huang, R.; Uribe-Romo, F. J.; Chae, H. K.; O'Keeffe, M.; Yaghi, O. M. Proc Natl Acad Sci U S A 2006, 103, 10186.
- Sing, K. S. W.; Everett, D. H.; Haul, R. A. W.; Moscou, L.; Pierotti, R. A.; Rouquerol, J.; Siemieniewska, T. Pure Appl. Chem. 1985, 57, 603.
- Horike, S.; Shimomura, S.; Kitagawa, S. Nat. Chem. 2009, 1, 695.
- Cooper, A. I. Adv. Mater. 2009, 21, 1291.
- Deng, H.; Grunder, S.; Cordova, K. E.; Valente, C.; Furukawa, H.; Hmadeh, M.; Gandara, F.; Whalley, A. C.; Liu, Z.; Asahina, S.; Kazumori, H.; O'Keeffe, M.; Terasaki, O.; Stoddart, J. F.; Yaghi, O. M. Science 2012, 336, 1018.
- Matsuda, R.; Kitaura, R.; Kitagawa, S.; Kubota, Y.; Belosludov, R. V.; Kobayashi, T. C.; Sakamoto, H.; Chiba, T.; Takata, M.; Kawazoe, Y.; Mita, Y. Nature 2005, 436, 238.
- Sumida, K.; Hill, M. R.; Horike, S.; Dailly, A.; Long, J. R. J. Am. Chem. Soc. 2009, 131, 15120.
- Zhao, X.; Wu, T.; Zheng, S. T.; Wang, L.; Bu, X.; Feng, P. Chem. Commun. 2011, 47, 5536.
- Go, Y. B.; Wang, X. Q.; Jacobson, A. J. Inorg. Chem. 2007, 46, 6594.
- Tsuruoka, T.; Furukawa, S.; Takashima, Y.; Yoshida, K.; Isoda, S.; Kitagawa, S. Angew. Chem. Int. Ed. 2009, 48, 4739.
- Kondo, M.; Okubo, T.; Asami, A.; Noro, S.; Yoshitomi, T.; Kitagawa, S.; Ishii, T.; Matsuzaka, H.; Seki, K. Angew. Chem. Int. Ed. 1999, 38, 140.
- Horike, S.; Tanaka, D.; Nakagawa, K.; Kitagawa, S. Chem. Commun. 2007, 3395.
- Tranchemontagne, D. J.; Hunt, J. R.; Yaghi, O. M. Tetrahedron 2008, 64, 8553.
- Hartmann, M.; Kunz, S.; Himsl, D.; Tangermann, O.; Ernst, S.; Wagener, A. Langmuir 2008, 24, 8634.
- Banerjee, R.; Phan, A.; Wang, B.; Knobler, C.; Furukawa, H.; O'Keeffe, M.; Yaghi, O. M. Science 2008, 319, 939.
- Sumida, K.; Horike, S.; Kaye, S. S.; Herm, Z. R.; Queen, W. L.; Brown, C. M.; Grandjean, F.; Long, G. J.; Dailly, A.; Long, J. R. Chem. Sci. 2010, 1, 184.
- Choi, J. S.; Son, W. J.; Kim, J.; Ahn, W. S. Micropor. Mesopor. Mater. 2008, 116, 727.
- (a) Tanaka, D.; Henke, A.; Albrecht, K.; Moeller, M.; Nakagawa, K.; Kitagawa, S.; Groll, J. Nat. Chem. 2010, 2, 410;(b) Hijikata, Y.; Horike, S.; Tanaka, D.; Groll, J.; Mizuno, M.; Kim, J.; Takata, M.; Kitagawa, S. Chem. Commun. 2011, 47, 7632.
- Beldon, P. J.; Fabian, L.; Stein, R. S.; Thirumurugan, A.; Cheetham, A. K.; Friscic, T. Angew. Chem. Int. Ed. 2010, 49, 9640.
- Friscic, T.; Reid, D. G.; Halasz, I.; Stein, R. S.; Dinnebier, R. E.; Duer, M. J. Angew. Chem. Int. Ed. 2010, 49, 712.
- (a) Sakamoto, H.; Kitaura, R.; Matsuda, R.; Kitagawa, S.; Kubota, Y.; Takata, M. Chem. Lett. 2010, 39, 218;(b) Matsuda, R.; Kitaura, R.; Kitagawa, S.; Kubota, Y.; Kobayashi, T. C.; Horike, S.; Takata, M. J. Am. Chem. Soc. 2004, 126, 14063.
- Chui, S. S. Y.; Lo, S. M. F.; Charmant, J. P. H.; Orpen, A. G.; Williams, I. D. Science 1999, 283, 1148.
- Diring, S.; Furukawa, S.; Takashima, Y.; Tsuruoka, T.; Kitagawa, S. Chem. Mater. 2010, 22, 4531.
- Mueller, U.; Schubert, M.; Teich, F.; Puetter, H.; Schierle-Arndt, K.; Pastre, J. J. Mater. Chem. 2006, 16, 626.
- Uemura, T.; Ono, Y.; Hijikata, Y.; Kitagawa, S. J. Am. Chem. Soc. 2010, 132, 4917.
- Hirai, K.; Furukawa, S.; Kondo, M.; Uehara, H.; Sakata, O.; Kitagawa, S. Angew. Chem. Int. Ed. 2011, 50, 8057.
- Dybtsev, D. N.; Chun, H.; Kim, K. Angew. Chem. Int. Ed. 2004, 43, 5033.
- Kaye, S. S.; Dailly, A.; Yaghi, O. M.; Long, J. R. J. Am. Chem. Soc. 2007, 129, 14176.
- Miyasaka, H.; Motokawa, N.; Chiyo, T.; Takemura, M.; Yamashita, M.; Sagayama, H.;Arima, T. H. J. Am. Chem. Soc. 2011, 133, 5338.
- Kawano, M.; Haneda, T.; Hashizume, D.; Izumi, F.; Fujita, M. Angew. Chem. Int. Ed. 2008, 47, 1269.
- Shekhah, O.; Liu, J.; Fischer, R. A.; Woll, C. Chem. Soc. Rev. 2011, 40, 1081.
- Uemura, K.; Kitagawa, S.; Fukui, K.; Saito, K. J. Am. Chem. Soc. 2004, 126, 3817.
- Meilikhov, M.; Yusenko, K.; Esken, D.; Turner, S.; Van Tendeloo, G.; Fischer, R. A. Eur. J. Inorg. Chem. 2010, 3701.
- Feldblyum, J. I.; Liu, M.; Gidley, D. W.; Matzger, A. J. J. Am. Chem. Soc. 2011, 133, 18257.
- Caskey, S. R.; Wong-Foy, A. G.; Matzger, A. J. J. Am. Chem. Soc. 2008, 130, 10870.
- Furukawa, H.; Ko, N.; Go, Y. B.; Aratani, N.; Choi, S. B.; Choi, E.; Yazaydin, A. O.; Snurr, R. Q.; O'Keeffe, M.; Kim, J.; Yaghi, O. M. Science 2010, 329, 424.
- McDonald, T. M.; D'Alessandro, D. M.; Krishna, R.; Long, J. R. Chem. Sci. 2011, 2, 2022.
- Li, D.; Kaneko, K. Chemical Physics Letters 2001, 335, 50.
- Ma, S. Q.; Sun, D. F.; Simmons, J. M.; Collier, C. D.; Yuan, D. Q.; Zhou, H. C. J. Am. Chem. Soc. 2008, 130, 1012.
- Britt, D.; Furukawa, H.; Wang, B.; Glover, T. G.; Yaghi, O. M. Proc. Natl. Acad. Sci. USA 2009, 106, 20637.
- Inubushi, Y.; Horike, S.; Fukushima, T.; Akiyama, G.; Matsuda, R.; Kitagawa, S. Chem. Commun. 2010, 46, 9229.
- Gucuyener, C.; van den Bergh, J.; Gascon, J.; Kapteijn, F. J. Am. Chem. Soc. 2010, 132, 17704.
- Murray, L. J.; Dinca, M.; Yano, J.; Chavan, S.; Bordiga, S.; Brown, C. M.; Long, J. R. J. Am. Chem. Soc. 2010, 132, 7856.
- Cho, S. H.; Ma, B. Q.; Nguyen, S. T.; Hupp, J. T.; Albrecht-Schmitt, T. E. Chem. Commun. 2006, 2563.
- (a) Seo, J. S.; Whang, D.; Lee, H.; Jun, S. I.; Oh, J.; Jeon, Y. J.; Kim, K. Nature 2000, 404, 982;(b) Wu, C. D.; Hu, A.; Zhang, L.; Lin, W. J. Am. Chem. Soc. 2005, 127, 8940.
続きを確認するには、ログインするか、新規登録が必要です。
アカウントをお持ちではありませんか?