
SAB2108553
Anti-FAH
IgG fraction of antiserum
About This Item
Empfohlene Produkte



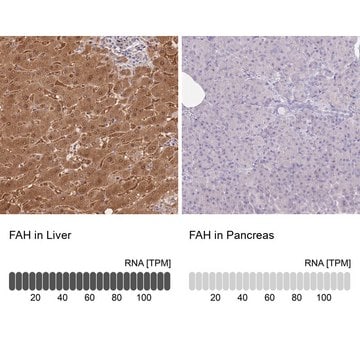

Biologische Quelle
rabbit
Qualitätsniveau
Konjugat
unconjugated
Antikörperform
IgG fraction of antiserum
Antikörper-Produkttyp
primary antibodies
Klon
polyclonal
Form
buffered aqueous solution
Mol-Gew.
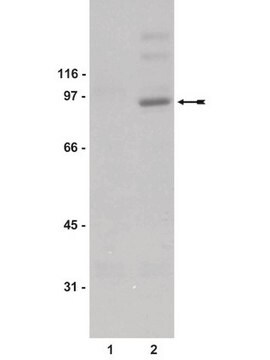
46 kDa
Speziesreaktivität
dog, horse, human, mouse, rat
Konzentration
0.5-1 mg/mL
Methode(n)
immunoblotting: suitable
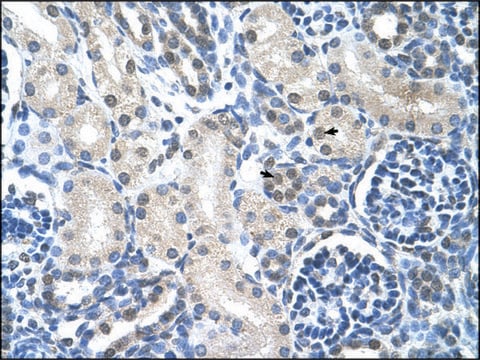
immunohistochemistry: suitable
Hinterlegungsnummer
NM_000137
UniProt-Hinterlegungsnummer
Versandbedingung
wet ice
Lagertemp.
−20°C
Posttranslationale Modifikation Target
unmodified
Angaben zum Gen
human ... FAH(2184)
Allgemeine Beschreibung
Immunogen
Biochem./physiol. Wirkung
Sequenz
Physikalische Form
Haftungsausschluss
Sie haben nicht das passende Produkt gefunden?
Probieren Sie unser Produkt-Auswahlhilfe. aus.
Lagerklassenschlüssel
10 - Combustible liquids
WGK
WGK 3
Flammpunkt (°F)
Not applicable
Flammpunkt (°C)
Not applicable
Analysenzertifikate (COA)
Suchen Sie nach Analysenzertifikate (COA), indem Sie die Lot-/Chargennummer des Produkts eingeben. Lot- und Chargennummern sind auf dem Produktetikett hinter den Wörtern ‘Lot’ oder ‘Batch’ (Lot oder Charge) zu finden.
Besitzen Sie dieses Produkt bereits?
In der Dokumentenbibliothek finden Sie die Dokumentation zu den Produkten, die Sie kürzlich erworben haben.
Unser Team von Wissenschaftlern verfügt über Erfahrung in allen Forschungsbereichen einschließlich Life Science, Materialwissenschaften, chemischer Synthese, Chromatographie, Analytik und vielen mehr..
Setzen Sie sich mit dem technischen Dienst in Verbindung.