
Oligonukleotydy antysensowne
Niniejszy artykuł podsumowuje kilka powszechnych mechanizmów modulacji genów antysensownych, a co ważniejsze, rozważania, które należy wziąć pod uwagę przy projektowaniu oligonukleotydu antysensownego (ASO). Po dziesięcioleciach badań nie ma twardych i szybkich zasad projektowania; nadal jest to metoda prób i błędów. Istnieją jednak wytyczne, których należy przestrzegać, aby proces ten był łatwiejszy do opanowania.
Przegląd sekcji
Pomoc przy projektowaniu
Potrzebujesz pomocy przy projektowaniu ASO lub nie możesz znaleźć modyfikacji w konfiguratorze zamówień online? Nasz zespół ds. usług technicznych może pomóc. Prosimy o wysłanie zapytania na adres [email protected].
Mechanizmy modulacji
Tradycyjna modulacja genów oparta na ASO (zwykle synonim wyciszania lub obniżania ekspresji genów, ale może być stosowana do poprawy ekspresji genów i, w co najmniej jednym konkretnym przypadku, wykazano, że prowadzi do zwiększenia ekspresji genów) jest ukierunkowana na mRNA i może mieć miejsce w jądrze lub cytoplazmie. W jądrze (pre-mRNA jest celem), modulacja zazwyczaj działa poprzez przekierowanie poliadenylacji1, zmianę zdarzeń splicingowych2 lub rozszczepianie wiązań międzynukleotydowych3, z których wszystkie występują podczas dojrzewania mRNA (Rysunek 1). W cytoplazmie (dojrzały mRNA jest celem) modulacja zazwyczaj działa albo poprzez zmianę translacji bez rozszczepienia4 lub rozszczepienie3, z których oba występują tuż przed / w trakcie translacji (Rysunek 2).
 do najbardziej wysuniętego na 3'- sygnał poliadenylacji na pre-mRNA i blokuje poliadenylację w tym miejscu, przekierowując ją w ten sposób do innego miejsca w górę łańcucha, co zwiększa ekspresję genu<sup>1</sup> B) do miejsca spliceosomu, zapobiegając w ten sposób prawidłowemu złożeniu spliceosomu, co prowadzi do pominięcia eksonu, a tym samym poprawy ekspresji genu chorobowego<sup>2</sup> (przez wielu nie jest uważana za prawdziwą ASO).sup>2</sup> (przez wielu nie uważane za prawdziwe ASO, są one często nazywane oligonukleotydami przełączającymi splice [SSO] lub bardziej ogólnie, oligonukleotydami blokującymi sterycznie [SBO]) C) ekson lub intron (w tym przypadku intron), prowadząc w ten sposób do rozszczepienia przez RNazę H<sup>3</sup>. W większości przypadków, mimo znacznej regulacji, wyciszenia lub zmiany, prawdopodobnie nastąpi pewne przetwarzanie niezmienionego pre-mRNA, a następnie eksport dojrzałego mRNA do cytoplazmy. S = sekwencja sygnałowa poliadenylacji (chociaż pokazano tu tylko jedną, może być więcej niż jedna na transkrypt).")
Rysunek 1. Mechanizmy modulacji genów w jądrze oparte na ASO.W przypadku ssaków, gDNA w jądrze jest transkrybowane do pre-mRNA. Egzogenna ASO w jądrze hybrydyzuje A) do najbardziej wysuniętego na 3'- sygnał poliadenylacji na pre-mRNA i blokuje poliadenylację w tym miejscu, przekierowując ją w ten sposób do innego miejsca w górę łańcucha, co zwiększa ekspresję genu1 B) do miejsca spliceosomu, zapobiegając w ten sposób prawidłowemu złożeniu spliceosomu, co prowadzi do pominięcia eksonu, a tym samym poprawy ekspresji genu chorobowego2 (przez wielu nie jest uważana za prawdziwą ASO).sup>2 (przez wielu nie uważane za prawdziwe ASO, są one często nazywane oligonukleotydami przełączającymi splice [SSO] lub bardziej ogólnie, oligonukleotydami blokującymi sterycznie [SBO]) C) ekson lub intron (w tym przypadku intron), prowadząc w ten sposób do rozszczepienia przez RNazę H3. W większości przypadków, mimo znacznej regulacji, wyciszenia lub zmiany, prawdopodobnie nastąpi pewne przetwarzanie niezmienionego pre-mRNA, a następnie eksport dojrzałego mRNA do cytoplazmy. S = sekwencja sygnałowa poliadenylacji (chociaż pokazano tu tylko jedną, może być więcej niż jedna na transkrypt).
 transkrybowane do pre-mRNA 2) pre-mRNA jest przetwarzane (dodawana jest 5' czapeczka i 3' ogon poli[A]) i splicingowane (usuwane są introny) w celu wytworzenia dojrzałego mRNA i 3) dojrzałe mRNA jest eksportowane do cytoplazmy. Egzogenny ASO hybrydyzuje do dojrzałego mRNA w cytoplazmie i wycisza (obniża) ekspresję genu poprzez A) zmianę translacji, w tym przypadku zahamowanie translacji poprzez zakłócenie montażu rybosomalnego na czapeczce 5'<sup>4</sup> (często nie jest to uważane za prawdziwe ASO, jest to przykład ogólnego SBO) lub B) rozszczepienie przez RNazę H (w szczególności RNazę H1 u ludzi)<sup>3</sup>. W większości przypadków, choć znacznie obniżona, translacja nadal występuje.")
Rysunek 2. Mechanizmy modulacji genów w cytoplazmie oparte na ASO.W przypadku ssaków, gDNA w jądrze jest 1) transkrybowane do pre-mRNA 2) pre-mRNA jest przetwarzane (dodawana jest 5' czapeczka i 3' ogon poli[A]) i splicingowane (usuwane są introny) w celu wytworzenia dojrzałego mRNA i 3) dojrzałe mRNA jest eksportowane do cytoplazmy. Egzogenny ASO hybrydyzuje do dojrzałego mRNA w cytoplazmie i wycisza (obniża) ekspresję genu poprzez A) zmianę translacji, w tym przypadku zahamowanie translacji poprzez zakłócenie montażu rybosomalnego na czapeczce 5'4 (często nie jest to uważane za prawdziwe ASO, jest to przykład ogólnego SBO) lub B) rozszczepienie przez RNazę H (w szczególności RNazę H1 u ludzi)3. W większości przypadków, choć znacznie obniżona, translacja nadal występuje.
ASO rozpoznają i hybrydyzują z docelowym mRNA poprzez parowanie zasad Watsona-Cricka. ASO, które prowadzą do rozszczepienia docelowego mRNA przez RNazę H (zarówno w jądrze, jak i cytoplazmie3) są szeroko badane w celach badawczych i terapeutycznych, a zatem są najlepiej poznane pod względem mechanizmu modulacji. Wykorzystując jony magnezu jako kofaktor, RNaza H (w szczególności RNaza H1 u ludzi) rozszczepia nić mRNA w heterodupleksie mRNA:DNA poprzez hydrolizę wiązania międzynukleotydowego (fosfodiestrowego)5. Po rozszczepieniu, ASO pozostaje nienaruszone, podczas gdy poprzednie wiązanie syzygijne jest teraz wolnymi grupami 3'-hydroksylowymi i 5'-fosforanowymi odpowiednio na fragmentach 5' i 3' zdegradowanego mRNA.
Rozważania projektowe
W zasadzie wyciszanie genów powinno być tak proste, jak wybranie sekwencji w docelowym mRNA; zamówienie komplementarnego ASO z parą zasad Watson-Crick od dostawcy; wprowadzenie go do badanego systemu (in vitro lub in vivo); i obserwowanie oczekiwanego efektu za pomocą odpowiedniego reportera. Istnieje jednak wiele czynników, które należy wziąć pod uwagę, aby pomyślnie zaprojektować ASO.
Miejsce hybrydyzacji
Zgodnie z zasadami parowania zasad Watson-Crick, ASO powinno hybrydyzować z dowolnym regionem docelowej sekwencji mRNA. Jednak mRNA składa się w struktury drugorzędowe, a nawet trzeciorzędowe, które prawdopodobnie blokują hybrydyzację ASO. Dlatego jako miejsca hybrydyzacji należy wybrać niefałdowane regiony mRNA. Istnieją metody mokrego laboratorium, takie jak mapowanie RNazy H, które są przydatne w przewidywaniu dostępnego miejsca6, ale dobrym miejscem do rozpoczęcia jest wypróbowanie algorytmu przewidywania fałdowania RNA, np. mfold.
Po zidentyfikowaniu regionu niefałdowanego, należy rozważyć, czy region ten służy jako miejsce wiązania dla spliceosomów, rybosomów, białek lub innych zespołów makromolekularnych. Historycznie, czapeczka 5', kodon inicjacyjny, 3' region nieulegający translacji / ogon polyA były dobrymi miejscami wyboru7. Nawet jeśli ASO nie aktywuje RNazy H, może nadal prowadzić do wyciszenia, ponieważ sterycznie zablokuje maszynerię potrzebną do dojrzewania lub translacji mRNA.
Degradacja przez nukleazy
In vivo i in vitro, wszystkie natywne ASO DNA szybko stają się bezużyteczne przez aktywność nukleazy. In vivo, chociaż zarówno endonukleazy, jak i egzonukleazy mogą prowadzić do degradacji8, egzonukleazy wydają się powodować większość uszkodzeń9,10. Aby być skutecznym, wszystkie ASO wymagają modyfikacji chemicznej, aby były odporne na degradację nukleazy. Chociaż dostępne są liczne analogi kwasów nukleinowych do modyfikowania ASO5,11, tutaj tylko te, które są częścią naszego standardowej oferty modyfikacji zostaną zbadane (Tabela 1).
Trzy regiony ASO podlegają modyfikacji (wiązania międzynukleotydowe, cukry i zasady), a we wszystkich kolejnych sekcjach modyfikacje są klasyfikowane zgodnie z ich podstawowym efektem, nawet jeśli kilka z nich ma więcej niż jeden efekt, np.np. modyfikacja X efekt pierwotny: poprawia powinowactwo wiązania; efekt wtórny: zmniejsza szkodliwy wpływ immunostymulacji (w tym artykule skupimy się na efekcie pierwotnym).
| Modyfikacja według typu | ||
|---|---|---|
Powiązania międzynukleotydowe | ||
| .Chemia | Skrót | Struktura |
| Phosphorothioate (aka Thiophosphate lub S-oligo) | PS (* w konstrukcjach sekwencyjnych) |  |
Cukry | ||
| Chemia. | Skrót | Struktura |
| Metyl RNA | 2'-OMe-RNA ([mA], [mC], [mG], & [mU] w konstruktach sekwencyjnych) | 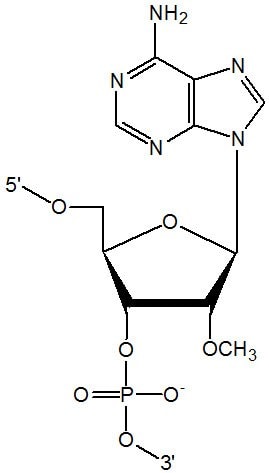 mA |
| Metoksyetylowy RNA | 2'-MOE-RNA ([moeA], [moe5C], [moeG], & [moe5U] w konstruktach sekwencyjnych) | 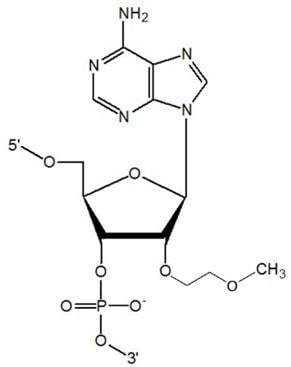 moeA |
| Fluoro RNA | 2'-F-RNA ([2flA], [2flC], [2flG], & [2flU] w konstruktach sekwencyjnych) |  2flA |
Phosphorothioate. Ta modyfikacja była jedną z niewielu, które są uważane za pierwszą generację. PS-ASO są odporne na nukleazy i dlatego mają dłuższy okres półtrwania w osoczu w porównaniu z natywnymi ASO DNA9. Ponadto zachowują one ujemne ładunki szkieletowe, co ułatwia wejście PS-ASO do komórki11. Co ciekawe, PS wydaje się mieć większy wpływ na transport i wejście do komórki niż na odporność na nukleazy12.
Jednak PS-ASO nie są całkowicie chronione przed nukleazami, mają zmniejszoną hybrydyzację do docelowych mRNA (patrz sekcja Powinowactwo wiązania) i muszą być stale podawane w dużych ilościach, aby utrzymać modulację11. Ponadto PS może wchodzić w interakcje z białkami in vivo, a tym samym prowadzić do negatywnych skutków ubocznych, w tym aktywności układu odpornościowego11.
Metyl RNA. Ta modyfikacja była jedną z niewielu, które są uważane za drugą generację. Stwierdzono, że w połączeniu z PS w ASO, 2'-OMe-RNA poprawia korzyści płynące z samego PS (tj. zwiększona odporność na nukleazy, okres półtrwania w osoczu i wychwyt tkankowy11).
Metoksyetyl RNA. Pochodna 2'-Me-RNA, 2'-O-metoksyetyl (2'-MOE) zyskała popularność w ostatnich latach po tym, jak została włączona do kilku zatwierdzonych leków23. Podobnie jak w przypadku innych modyfikacji 2' rybonukleotydów, 2'-MOE skutkuje zwiększoną stabilnością dupleksu w połączeniu z celami RNA.
Fluoro RNA. Zastąpienie grupy 2′-hydroksylowej w RNA fluorem znacznie zwiększa jego temperaturę topnienia, stabilność chemiczną i odporność na nukleazy.
Immunostymulacja
Bakteryjne DNA zawiera znacznie wyższą częstotliwość dinukleotydów CpG (wiązanie cytozyna-fosfodiester-guanina) pozbawionych metylacji niż DNA kręgowców. Dzieje się tak głównie dlatego, że dinukleotydy CpG są niedostatecznie reprezentowane w genomie kręgowców, a 80% z nich jest znakowanych grupami metylowymi13.Ponieważ motyw CpG w bakteriach wyzwala aktywację komórek B, komórek NK, monocytów i cytokin, podczas gdy motyw CpG kręgowców nie, jest to prawdopodobnie przynajmniej jeden ze sposobów, w jaki układ odpornościowy rozpoznaje infekcję bakteryjną13. ASO zawierające niemetylowane motywy CpG (CpsG: wiązanie cytozyna-fosforotioan-guanina jest jeszcze silniejsze14) stymulują układ odpornościowy w sposób podobny do bakteryjnego DNA i mogą być odpowiedzialne za niektóre zgłaszane efekty z wczesnych badań antysensownych.
Aby uniknąć immunostymulacji, projektuj ASO pozbawione motywów CpG / CpsG, jeśli to możliwe, lub przynajmniej te pozbawione następującego rozszerzonego motywu, który wywołuje najsilniejszą odpowiedź immunologiczną14:
- puryna-puryna-CpG-pirymidyna-pirydmidyna
Zważywszy, że może to być trudne do uniknięcia ze względu na komplementarny charakter sekwencji wyboru miejsca docelowego, następnym najlepszym krokiem jest zastąpienie cytozyny w CpG / CpsG przez 5'-metylocytozyną (Tabela 2), która, jak wykazano, znacznie zmniejsza immunostymulację15.
| Modyfikacja według typu | ||
|---|---|---|
Zasady | ||
| .Chemia | Skrót | Struktura |
| 5-metylocytozyna | 5-Me-dC ([5MedC] w konstruktach sekwencyjnych) | 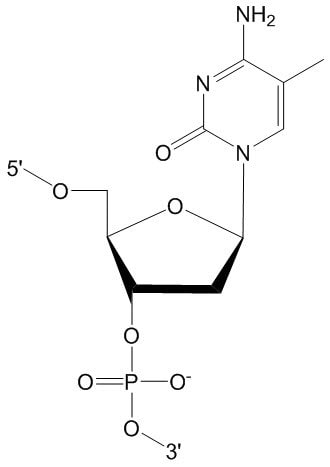 |
Długość sekwencji
Optymalne długości wynoszą zwykle od 12 do 28 zasad6. Sekwencje krótsze niż 12 zasad zwiększają prawdopodobieństwo hybrydyzacji poza miejscem docelowym, podczas gdy sekwencje dłuższe niż 25 zasad zwiększają prawdopodobieństwo zmniejszonego wychwytu komórkowego6.
Samokomplementarność
ASO należy sprawdzić pod kątem struktury drugorzędowej i tworzenia dimerów oligonukleotydów, ponieważ każdy z nich może zakłócać hybrydyzację z sekwencją miejsca docelowego. Jeśli to możliwe, zaprojektuj ASO tak, aby miał jak najsłabszą strukturę drugorzędową, a także nie tworzył dimerów. Nasz kalkulator sekwencji oligonukleotydów OligoEvaluator™ pozwala na szybkie określenie tych samoformujących się struktur.
Struktury kwartetu G
ASO zawierające odcinki dwóch lub więcej nukleotydów C lub G są w stanie tworzyć nietypowe struktury, które mogą powodować niepożądane efekty poza celem. Najbardziej powszechne i badane są odcinki zasad G, które mogą prowadzić do tworzenia kwartetów G16. Wykazano, że kwartety te wiążą się z białkami, w tym czynnikami transkrypcyjnymi17, które mogą naśladować, a tym samym zakłócać aktywność antysensowną.
Aby uniknąć tworzenia tych kwartetów, projektuj ASO bez tych odcinków polyG, jeśli to możliwe. Ponownie, biorąc pod uwagę, że może to być niewykonalne, następnym najlepszym krokiem jest zastąpienie guaniny przez 7-deaza-dG (Tabela 3), co zablokuje tworzenie się kwartetu18.
W celu uniknięcia tworzenia się tych kwartetów, należy zaprojektować ASO pozbawione tych odcinków polyG, jeśli to możliwe.
| Modyfikacja według typu | ||
|---|---|---|
Bazy | ||
| Chemia | Skrót | Struktura |
| 7-deaza-dG | [Deaza-dG] w konstrukcjach sekwencyjnych |  |
Motywy funkcjonalne
Analiza statystyczna eksperymentów PS-ASO wykazała, że następujące motywy:
- CCAC
- TCCC
- ACTC
- GCCA
- CTCT
korelują ze zwiększoną skutecznością antysensu, podczas gdy te motywy:
- GGG
- ACTG
- AAA
- TAA
zmniejszają aktywność antysensowną19. Stwierdzono, że aktywność RNazy H jest niezależna od sekwencji20; Dlatego uważa się, że motywy wzmacniające prowadzą do zwiększonej stabilności termodynamicznej heterodupleksu mRNA:ASO poprzez przewagę parowania zasad GC Watson-Crick.
Przywiązanie
Jak już wspomniano, niezwykle ważne jest zidentyfikowanie miejsca w docelowym mRNA, które jest wolne od fałd, a także zapewnienie, że ASO nie ma również szkodliwej samouzupełniania. Jednak same te rozważania nie wystarczą, aby zapewnić prawidłową hybrydyzację. Różne czynniki, takie jak PS, mogą zmniejszać powinowactwo wiązania ASO do miejsca docelowego11, co z kolei minimalizuje skuteczność antysensu.
Stwierdzono, że modyfikacje ASO trzeciej generacji są nie tylko odporne na nukleazy, ale także poprawiają powinowactwo wiązania. Zablokowany kwas nukleinowy (Tabela 4), z jego ograniczoną strukturą pierścieniową, jest szczególnie przydatny do poprawy powinowactwa wiązania ASO i skuteczności (zmiana temperatury topnienia na dodanie monomeru waha się od +3 do +11 °C w porównaniu tylko do natywnego DNA21).
| Modyfikacja według typu | ||
|---|---|---|
Zasady | ||
| .Chemia | Skrót | Struktura |
| Zablokowany kwas nukleinowy | Zablokowana zasada ([+A], [+C], [+G], & [+T] w konstrukcjach sekwencyjnych) |  |
The Construct
Aby dać wgląd w sekwencje ASO, podano tutaj przykłady kilku leków antysensownych (często główny cel prowadzenia badań nad antysensem), które zostały zatwierdzone lub są w trakcie badań klinicznych. Leki te są przykładami (lub oczekuje się, że będą w przypadku tych w badaniach klinicznych) wszystkich pożądanych rezultatów, jeśli chodzi o antysens: dobrego projektu, dostępnego mechanizmu dostarczania i skutecznej modulacji. Te same wyniki mają kluczowe znaczenie dla powodzenia eksperymentów badawczych (nasze ASO są przeznaczone do badań in vitro i in vivo na zwierzętach RUO [Research Use Only]).
Pierwsza generacja. W 1998 r. Fomivirsen (nazwa handlowa Vitravene) był pierwszym zatwierdzonym lekiem antysensownym. Był on stosowany w leczeniu cytomegalowirusowego zapalenia siatkówki (CMV) u pacjentów z obniżoną odpornością, w tym chorych na AIDS. Lek był dostarczany przez wstrzyknięcie do ciała szklistego. 21-merowa ASO ze wszystkimi wiązaniami międzynukleotydowymi PS ma następującą sekwencję:
- G*C*G*T*T*G*C*T*C*T*C*T*T*C*T*T*G*C*G
○ * = PS
i działa poprzez hamowanie translacji transkrybowanego mRNA z genu CMV UL12322. Ostatecznie został wycofany z rynku, ponieważ rozwój HAART (wysoce aktywnej terapii antyretrowirusowej) w leczeniu HIV zmniejszył liczbę przypadków CMV o 75% i dlatego doprowadził do słabej sprzedaży.
Ponieważ ASO oparte wyłącznie na PS nie są całkowicie chronione przed nukleazami, mają zmniejszoną hybrydyzację z docelowym mRNA, muszą być stale podawane w dużych ilościach w celu utrzymania modulacji i mogą wchodzić w interakcje z białkami, co może prowadzić do negatywnych skutków ubocznych11, konstrukty pierwszej generacji zostały w dużej mierze porzucone w procesach badawczo-rozwojowych.
Druga generacja. W 2013 roku Mipomersen (nazwa handlowa Kynamro®) stał się drugim zatwierdzonym lekiem antysensownym. Jest on stosowany w leczeniu rodzinnej hipercholesterolemii, dziedzicznego zaburzenia. Lek jest dostarczany przez wstrzyknięcie podskórne. 20-merowa ASO ze wszystkimi wiązaniami międzynukleotydowymi PS ma następującą sekwencję:
- G*mC*mU*mC*A*G*T*mC*T*G*mC*T*mC*G*mC*A*mC*mC
○ Podkreślenie = 2'-O-MOE-RNA (MOE to 2-metoksyetyl)
.nbsp; ○ m = metyl, tj.e. 5-Me-dC & 5-Me-U
○ * = PS
i działa poprzez hamowanie translacji mRNA apolipoproteiny B-10023. Istnieje ryzyko poważnego uszkodzenia wątroby, więc lek musi być częścią planu zarządzania ryzykiem.
Cząsteczki antysensowne drugiej generacji, takie jak Mipomersen, są zaprojektowane z konfiguracją 5-10-5 gapmer24. Można to zobaczyć w powyższej sekwencji: 5' i 3' skrzydełka po 5 zasad (zmodyfikowanych cukrem o zwiększonym powinowactwie do wiązania i odpornych na nukleazę) i centralna przerwa 10 standardowych dezoksyrybonukleotydów (bez modyfikacji cukrem), która umożliwia wiązanie RNazy H11.
W tym konkretnym przypadku skrzydła składają się z 2'-O-MOE-RNA (MOE to 2-metoksyetyl), odmiany modyfikacji 2'-O-metylowej RNA.
Trzecia generacja. Od 2017 r. Miravirsen (SPC3649) znajduje się w II fazie badań klinicznych25. Jest on testowany jako lek na wirusowe zapalenie wątroby typu C (HCV). Lek jest dostarczany przez wstrzyknięcie podskórne. 15-merowa ASO ze wszystkimi wiązaniami międzynukleotydowymi PS ma następującą sekwencję26:
- C*C*A*T*G*T*C*A*C*A*.C*T*C*C
○ Underline = LNA
&○ * = PS
i działa poprzez hybrydyzację z ludzkim miRNA, miR-122. Zapobiega to przenoszeniu przez miR-122 argonauty do regionu 5'-UTR RNA wirusa HCV, gdzie normalnie się wiąże, a tym samym chroni przed degradacją przez nukleazy25. Dlatego Miravirsen pozwala na zniszczenie wirusowego RNA.
Chociaż Miravirsen nie jest tradycyjnym ASO, ponieważ jest ukierunkowany na miRNA, a zatem tylko pośrednio prowadzi do degradacji mRNA, jest to jeden z najlepszych przykładów konstruktu trzeciej generacji zawierającego LNA, dlatego został tutaj uwzględniony.
Target Check
Końcowa niezmodyfikowana sekwencja ASO powinna zostać poddana testowi BLAST aby upewnić się, że jakakolwiek hybrydyzacja poza celem - najlepiej żadna - nie będzie zakłócać aktywności antysensownej lub prowadzić do niedopuszczalnej toksyczności.
Uwagi dotyczące jakości
Do doświadczeń in vivo na zwierzętach zalecamy ASO. na zwierzętach, zalecamy, aby ASO były poddawane oczyszczaniu in vivo z wymianą soli (zastępuje toksyczne jony amonowe z chemii syntezy fosforoamidytów fizjologicznymi jonami sodu), testowanie endotoksyn (zapewnia obecność pirogenów poniżej dopuszczalnego pułapu) i filtrację (zmniejsza liczbę zanieczyszczających CFU poniżej dopuszczalnego pułapu). Nasz produkt iScale Oligos™ to większe ilości materiału do projektów in vivo, które można zamówić z tym oczyszczaniem i wszystkimi tymi dodatkowymi usługami.
Dostarczanie & Toksyczność
Chociaż wykracza to poza zakres tego artykułu, istnieje kilka doskonałych artykułów przeglądowych, które omawiają różne mechanizmy dostarczania, a także potencjalne toksyczności11,27,28,29,30.
Wnioski
Kiedy zaprojektowałeś ASO, które chcesz wypróbować w eksperymencie, jesteśmy gotowi zsyntetyzować je dla Ciebie (nasze ASO są przeznaczone do badań in vitro i in vivo na zwierzętach (RUO [Research Use Only]). Jeśli potrzebna jest dodatkowa pomoc, zwłaszcza w zakresie możliwości produkcji ASO z niestandardowymi modyfikacjami, prosimy o przesłanie zapytania na adres [email protected].
.Referencje
Zaloguj się lub utwórz konto, aby kontynuować.
Nie masz konta użytkownika?Dla wygody naszych klientów ta strona została przetłumaczona maszynowo. Dołożyliśmy starań, aby zapewnić dokładne tłumaczenie maszynowe. Tłumaczenie maszynowe nie jest jednak doskonałe. Jeśli tłumaczenie maszynowe nie spełnia Twoich oczekiwań, przejdź do wersji w języku angielskim.