Journal of inherited metabolic disease, 33(3), 211-222 (2010-05-06)
Mitochondrial dysfunction and oxidative stress are central to the molecular basis of several human diseases associated with neuromuscular disabilities. We hypothesize that mitochondrial dysfunction also contributes to the neuromuscular symptoms observed in patients with ethylmalonic aciduria and homozygosity for ACADS
Ethylmalonic encephalopathy (EE) is a rare autosomal recessive disorder caused by mutations in the ETHE1 gene and characterized by chronic diarrhea, encephalopathy, relapsing petechiae and acrocyanosis. Nephrotic syndrome has been described in an infant with EE but the renal histology
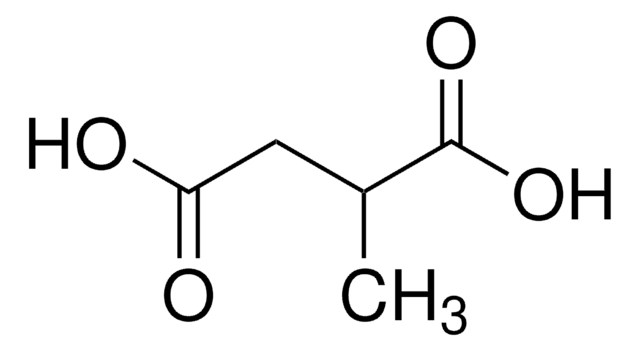
High concentrations of ethylmalonic acid (EMA) are found in tissues and biological fluids of patients affected by ethylmalonic encephalopathy (EE), as well as by deficiency of short-chain acyl-CoA dehydrogenase (SCAD) activity and other illnesses characterized by developmental delay and other
Short-chain acyl-CoA dehydrogenase deficiency (SCADD) is an inborn error, biochemically characterized by increased plasma butyrylcarnitine (C4-C) concentration and increased ethylmalonic acid (EMA) excretion and caused by rare mutations and/or common gene variants in the SCAD encoding gene. Although its clinical
Toxicological sciences : an official journal of the Society of Toxicology, 129(2), 268-279 (2012-07-24)
Ibipinabant (IBI), a potent cannabinoid-1 receptor (CB1R) antagonist, previously in development for the treatment of obesity, causes skeletal and cardiac myopathy in beagle dogs. This toxicity was characterized by increases in muscle-derived enzyme activity in serum and microscopic striated muscle
Nuestro equipo de científicos tiene experiencia en todas las áreas de investigación: Ciencias de la vida, Ciencia de los materiales, Síntesis química, Cromatografía, Analítica y muchas otras.