
MABN2427
Anti-PolyQ-spezifischer Antikörper, Klon MW1
clone MW1, from mouse
Synonym(e):
Htt mutant, PolyQ-Htt, Clone MW1, Atrophin-1
About This Item
Empfohlene Produkte




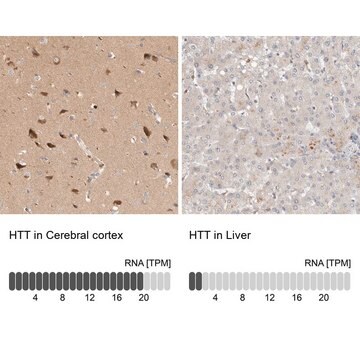

Biologische Quelle
mouse
Qualitätsniveau
Antikörperform
purified immunoglobulin
Antikörper-Produkttyp
primary antibodies
Klon
MW1, monoclonal
Speziesreaktivität
mouse, rat, human
Verpackung
antibody small pack of 25 μg
Methode(n)
immunocytochemistry: suitable
immunohistochemistry: suitable
western blot: suitable
Isotyp
IgG2b
NCBI-Hinterlegungsnummer
UniProt-Hinterlegungsnummer
Versandbedingung
ambient
Posttranslationale Modifikation Target
unmodified
Angaben zum Gen
human ... ATN1(1822)
Verwandte Kategorien
Allgemeine Beschreibung
Spezifität
Immunogen
Anwendung
Neurowissenschaft
Immunhistochemische Analyse: Mit einer repräsentativen Charge wurden erweiterte polyQ-Repeats in immunhistochemischen Anwendungen nachgewiesen (Ko, J., et. al. (2001). Brain Res Bull. 56(3-4):319-29).
Immunzytochemische Analyse: Mit einer repräsentativen Charge wurden erweiterte polyQ-Repeats in immunzytochemischen Anwendungen nachgewiesen (Legleiter, J., et. al. (2009). J Biol Chem. 284(32):21647-58).
Qualität
Western-Blot-Analyse: Mit 0,5 µg/ml dieses Antikörpers wurden erweiterte polyQ-Repeats in 10 µg Hirngewebelysat der Maus nachgewiesen. Es reagiert nicht mit normalen Htt.
Zielbeschreibung
Physikalische Form
Lagerung und Haltbarkeit
Sonstige Hinweise
Haftungsausschluss
Not finding the right product?
Try our Produkt-Auswahlhilfe.
WGK
WGK 1
Analysenzertifikate (COA)
Suchen Sie nach Analysenzertifikate (COA), indem Sie die Lot-/Chargennummer des Produkts eingeben. Lot- und Chargennummern sind auf dem Produktetikett hinter den Wörtern ‘Lot’ oder ‘Batch’ (Lot oder Charge) zu finden.
Besitzen Sie dieses Produkt bereits?
In der Dokumentenbibliothek finden Sie die Dokumentation zu den Produkten, die Sie kürzlich erworben haben.
Unser Team von Wissenschaftlern verfügt über Erfahrung in allen Forschungsbereichen einschließlich Life Science, Materialwissenschaften, chemischer Synthese, Chromatographie, Analytik und vielen mehr..
Setzen Sie sich mit dem technischen Dienst in Verbindung.