Journal of inherited metabolic disease, 15(2), 204-212 (1992-01-01)
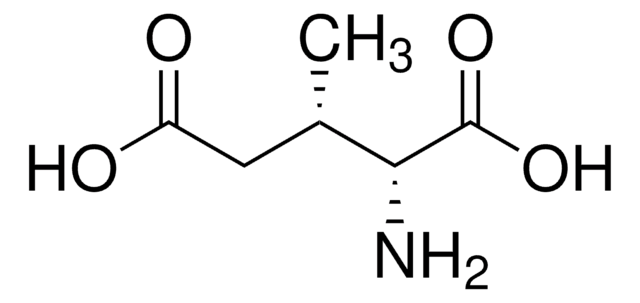
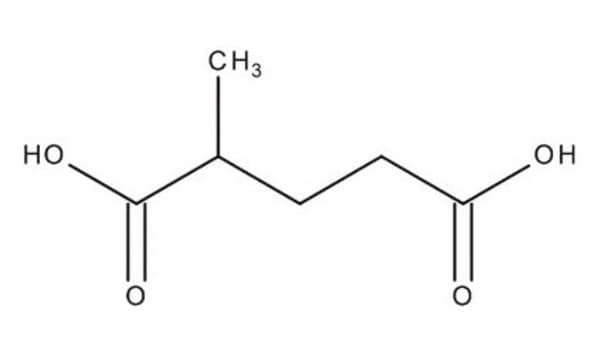
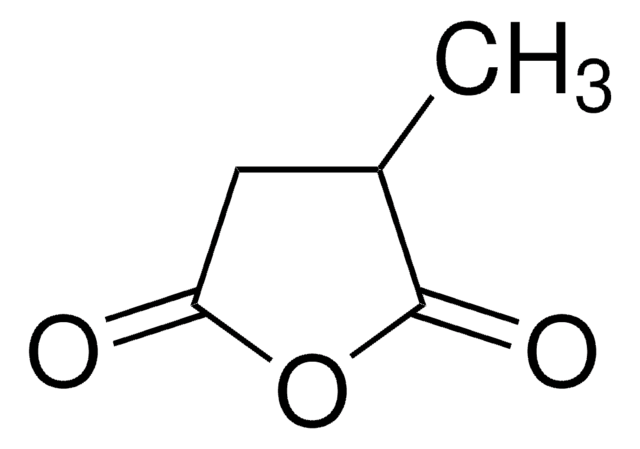
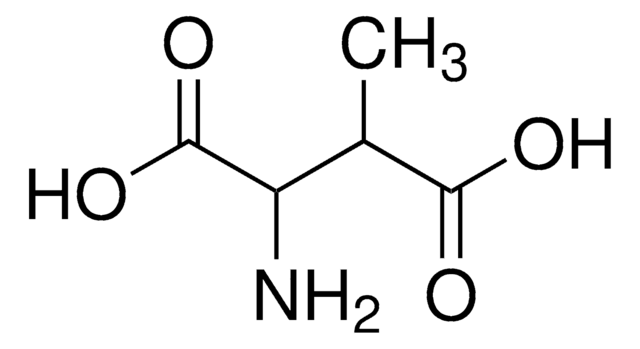
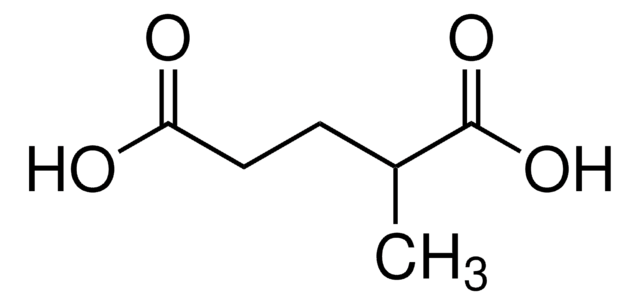
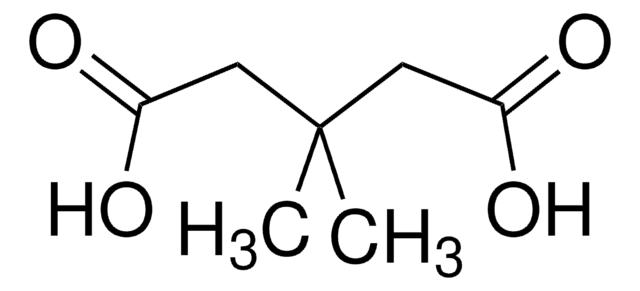
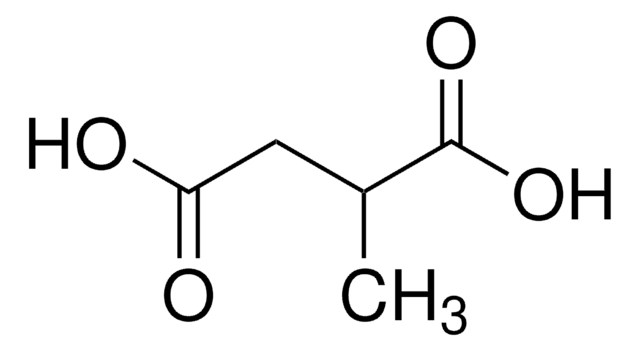
The Mendelian disorder known as 3-methylgutaconic aciduria (McKusick 250950) gives evidence of allelic and locus heterogeneity. Type 1 has a mild clinical phenotype and confirmed 3-methylgutaconyl-CoA hydratase deficiency; inheritance is autosomal recessive. Other forms have major clinical manifestations and subdivide
The Journal of pediatrics, 119(5), 738-747 (1991-11-01)
Seven boys with an apparently X-linked syndrome of dilated cardiomyopathy, growth retardation, neutropenia, and persistently elevated urinary levels of 3-methylglutaconate, 3-methylglutarate, and 2-ethylhydracrylate were studied. The natural history of the disorder was characterized by severe or lethal cardiac disease and
European journal of pediatrics, 146(5), 484-488 (1987-09-01)
Persistent excretion of 3-methylglutaconic acid was found in a 6-month-old infant with multiple minor physical malformations and delayed development. During two episodes of intercurrent viral illness, the patient developed severe metabolic acidosis and excreted large amounts of lactate, 3-hydroxybutyrate and
Notre équipe de scientifiques dispose d'une expérience dans tous les secteurs de la recherche, notamment en sciences de la vie, science des matériaux, synthèse chimique, chromatographie, analyse et dans de nombreux autres domaines..