
43659
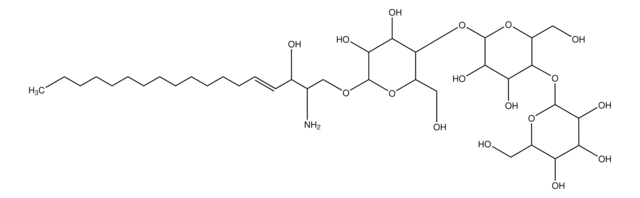
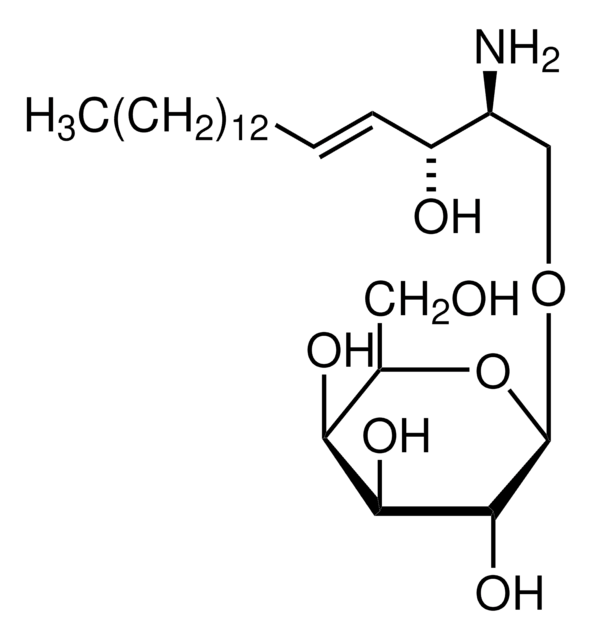
Glucosylsphingosine
≥98.0% (TLC)
Sinónimos:
(2S,3R,4E)-2-Amino-3-hydroxy-4-octadecen-1-yl β-D-glucopyranoside, 1-β-D-Glucosylsphingosine, Glucosyl-C18-sphingosine
About This Item
Productos recomendados








Quality Level
assay
≥98.0% (TLC)
form
powder
lipid type
sphingolipids
storage temp.
−20°C
SMILES string
O[C@@H]1[C@@H](O)[C@H](OC[C@H](N)[C@H](O)/C=C/CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O
InChI
1S/C24H47NO7/c1-2-3-4-5-6-7-8-9-10-11-12-13-14-15-19(27)18(25)17-31-24-23(30)22(29)21(28)20(16-26)32-24/h14-15,18-24,26-30H,2-13,16-17,25H2,1H3/b15-14+/t18-,19+,20?,21+,22+,23?,24+/m0/s1
InChI key
HHJTWTPUPVQKNA-JLRUQHRASA-N
Biochem/physiol Actions
Storage Class
11 - Combustible Solids
wgk_germany
WGK 3
flash_point_f
Not applicable
flash_point_c
Not applicable
Certificados de análisis (COA)
Busque Certificados de análisis (COA) introduciendo el número de lote del producto. Los números de lote se encuentran en la etiqueta del producto después de las palabras «Lot» o «Batch»
¿Ya tiene este producto?
Encuentre la documentación para los productos que ha comprado recientemente en la Biblioteca de documentos.
Nuestro equipo de científicos tiene experiencia en todas las áreas de investigación: Ciencias de la vida, Ciencia de los materiales, Síntesis química, Cromatografía, Analítica y muchas otras.
Póngase en contacto con el Servicio técnico