
推薦產品



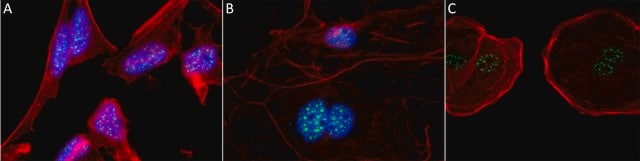
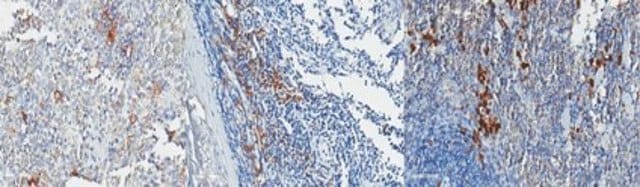
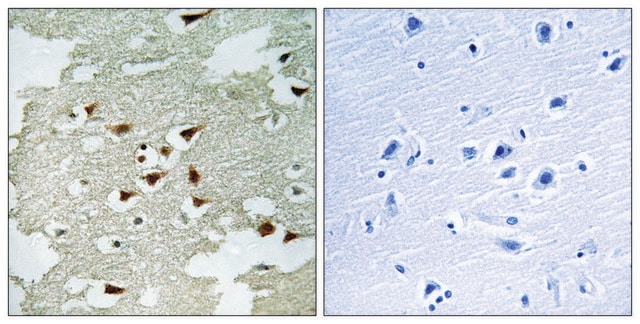

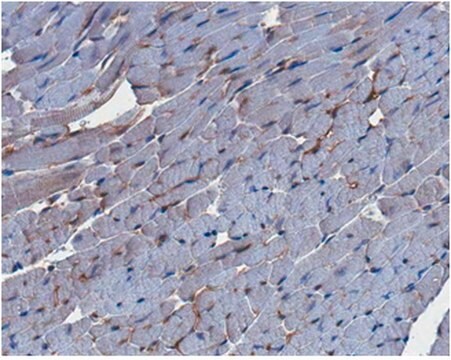


生物源
rabbit
品質等級
共軛
unconjugated
抗體表格
affinity isolated antibody
抗體產品種類
primary antibodies
無性繁殖
polyclonal
形狀
buffered aqueous glycerol solution
物種活性
human
技術
immunoblotting: 0.04-0.4 μg/mL
immunofluorescence: 0.25-2 μg/mL
immunohistochemistry: 1:500-1:1000
免疫原序列
DDGVQFHAFGRVLSGTIHAGQPVKVLGENYTLEDEEDSQICTVGRLWISVARYHIEVNRVPAGNWVLIEGVDQPIVKTATITEPRGNEEAQIFRPLKFNTTSVIKIAVEPV
UniProt登錄號
運輸包裝
wet ice
儲存溫度
−20°C
目標翻譯後修改
unmodified
基因資訊
human ... EFTUD2(9343)
一般說明
EFTUD2 (Elongation factor Tu GTP binding domain containing 2) is a member of the U5 small nuclear ribonucleoprotein particle (snRNP) located on chromosome region 17q21.31. It encodes a small GTPase (116kDa) component of the major spliceosome. It consists of a GTP-binding domain and several other conserved domains homologous to the translational elongation factor EF-2. Its N-terminal domain has an acidic domain.
免疫原
116 kDa U5 small nuclear ribonucleoprotein component recombinant protein epitope signature tag (PrEST)
應用
All Prestige Antibodies Powered by Atlas Antibodies are developed and validated by the Human Protein Atlas (HPA) project and as a result, are supported by the most extensive characterization in the industry.
The Human Protein Atlas project can be subdivided into three efforts: Human Tissue Atlas, Cancer Atlas, and Human Cell Atlas. The antibodies that have been generated in support of the Tissue and Cancer Atlas projects have been tested by immunohistochemistry against hundreds of normal and disease tissues and through the recent efforts of the Human Cell Atlas project, many have been characterized by immunofluorescence to map the human proteome not only at the tissue level but now at the subcellular level. These images and the collection of this vast data set can be viewed on the Human Protein Atlas (HPA) site by clicking on the Image Gallery link. We also provide Prestige Antibodies® protocols and other useful information.
The Human Protein Atlas project can be subdivided into three efforts: Human Tissue Atlas, Cancer Atlas, and Human Cell Atlas. The antibodies that have been generated in support of the Tissue and Cancer Atlas projects have been tested by immunohistochemistry against hundreds of normal and disease tissues and through the recent efforts of the Human Cell Atlas project, many have been characterized by immunofluorescence to map the human proteome not only at the tissue level but now at the subcellular level. These images and the collection of this vast data set can be viewed on the Human Protein Atlas (HPA) site by clicking on the Image Gallery link. We also provide Prestige Antibodies® protocols and other useful information.
生化/生理作用
EFTUD2 (Elongation factor Tu GTP binding domain containing 2) acts as a component of the spliceosome complex during translocation of mRNA on the ribosome. It produces mature mRNAs by binding to GTP during the processing of precursor mRNAs. Haploinsufficient genetic mutation in EFTUD2 causes a sporadic malformation syndrome, mandibulofacial dysostosis with microcephaly (MFDM), characterized with severe craniofacial abnormalities, microcephaly, growth delay, hearing loss, cleft palate, choanal atresia and dysmorphic features.
特點和優勢
Prestige Antibodies® are highly characterized and extensively validated antibodies with the added benefit of all available characterization data for each target being accessible via the Human Protein Atlas portal linked just below the product name at the top of this page. The uniqueness and low cross-reactivity of the Prestige Antibodies® to other proteins are due to a thorough selection of antigen regions, affinity purification, and stringent selection. Prestige antigen controls are available for every corresponding Prestige Antibody and can be found in the linkage section.
Every Prestige Antibody is tested in the following ways:
Every Prestige Antibody is tested in the following ways:
- IHC tissue array of 44 normal human tissues and 20 of the most common cancer type tissues.
- Protein array of 364 human recombinant protein fragments.
聯結
Corresponding Antigen APREST75576
外觀
Solution in phosphate-buffered saline, pH 7.2, containing 40% glycerol and 0.02% sodium azide
法律資訊
Prestige Antibodies is a registered trademark of Merck KGaA, Darmstadt, Germany
免責聲明
Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to humans or animals.
未找到適合的產品?
試用我們的產品選擇工具.
儲存類別代碼
10 - Combustible liquids
水污染物質分類(WGK)
WGK 1
閃點(°F)
Not applicable
閃點(°C)
Not applicable
Lei Lei et al.
Nucleic acids research, 45(6), 3422-3436 (2016-12-03)
Haploinsufficiency of EFTUD2 (Elongation Factor Tu GTP Binding Domain Containing 2) is linked to human mandibulofacial dysostosis, Guion-Almeida type (MFDGA), but the underlying cellular and molecular mechanisms remain to be addressed. We report here the isolation, cloning and functional analysis
T Achsel et al.
Molecular and cellular biology, 18(11), 6756-6766 (1998-10-17)
The human small nuclear ribonucleoprotein (snRNP) U5 is biochemically the most complex of the snRNP particles, containing not only the Sm core proteins but also 10 particle-specific proteins. Several of these proteins have sequence motifs which suggest that they participate
Margaret P Adam et al.
GeneReviews(?), 2014 Jul 03 (2014-07-08)
Mandibulofacial dysostosis with microcephaly (MFDM) is characterized by malar and mandibular hypoplasia; microcephaly (congenital or postnatal onset); malformations of the pinna, auditory canal, and/or middle ear (ossicles and semi-circular canals) with associated conductive hearing loss; and distinctive facial features (metopic
Daphné Lehalle et al.
Human mutation, 35(4), 478-485 (2014-01-29)
Mandibulofacial dysostosis, Guion-Almeida type (MFDGA) is a recently delineated multiple congenital anomalies/mental retardation syndrome characterized by the association of mandibulofacial dysostosis (MFD) with external ear malformations, hearing loss, cleft palate, choanal atresia, microcephaly, intellectual disability, oesophageal atresia (OA), congenital heart
S K Gandomi et al.
Clinical genetics, 87(1), 80-84 (2013-11-26)
Mandibulofacial dysostosis with microcephaly (MFDM) is a sporadic malformation syndrome with severe craniofacial abnormalities, microcephaly, developmental delay, and dysmorphic features. Most cases of clinically diagnosed MFDM remain genetically unexplained, and to the best of our knowledge a total of 35
我們的科學家團隊在所有研究領域都有豐富的經驗,包括生命科學、材料科學、化學合成、色譜、分析等.
聯絡技術服務