Description générale
La maladie de Huntington (MH [chorée de Huntington]) appartient à une famille de maladies polyglutaminiques, qui comprend l'atrophie dentato-rubro-pallido-luysienne (ADRPL), l'atrophie musculaire spino-bulbaire (SMA [Maladie de Kennedy]) et l'ataxie spinocérébelleuse (SCA) de types 1, 2, 3, 6, 7 et 17. Dans ces maladies, les allèles non pathogènes contiennent moins de 35 répétitions consécutives de glutamine et codent un domaine de polyglutamine normal. En revanche, les allèles pathogènes contiennent habituellement 39 répétitions consécutives ou plus de glutamine. Des nombres de répétition plus élevés conduisent à des âges d'apparition plus bas. Les patients présentant 40 à 60 répétitions de glutamine développent normalement une maladie à l'âge adulte, tandis que les patients avec plus de 60 répétitions développent une maladie juvénile. Chaque trouble d'expansion de la polyglutamine présente une pathologie caractéristique, avec une perte neuronale évidente dans des régions spécifiques du cerveau. La MH résulte d'expansions à partir d'un tractus de glutamine dans une protéine cytosolique de grande taille connue sous le nom de huntingtine.
Spécificité
L'épitope de l'anticorps MAB1574 s'est avéré être une séquence homopolymérique de glutamines. L'immunogène d'origine était la protéine de liaison à la boîte TATA (TATA Box-binding protein ou TBP), un facteur de transcription général contenant une succession de 38 glutamines (Lescure et al). D'autres protéines contenant de la polyglutamine sont reconnues par le MAB1574, notamment celles impliquées dans plusieurs maladies neurodégénératives humaines causées par une expansion répétée de CAG, comme la maladie de Huntington et l'ataxie spinocérébelleuse de type 2, 3 et 7 (Trottier et al., 1995). Il est important de noter que, pour les protéines impliquées dans ces troubles neurodégénératifs, le MAB1574 a montré des propriétés de détection remarquables, les protéines pathologiques contenant une expansion de polyglutamine (37 glutamines) étant beaucoup mieux détectées que les protéines de type sauvage (Trottier et al., 1995). Le MAB1574 a été utilisé pour identifier de nouvelles maladies neurodégénératives causées par une expansion de polyglutamine, ainsi que pour aider au clonage des gènes affectés correspondants (Trottier 1995-1998 ; Imbert 1996 ; Stevanin 1996). Le MAB1574 est également capable de détecter les inclusions intracellulaires, une autre caractéristique de ces maladies (Paulson, 1997).
Immunogène
Région N-terminale de la protéine de liaison à la boîte TATA (TBP) humaine.
Application
Domaine de recherche
Neurosciences
L'anticorps anti-Polyglutamine-Expansion Diseases Marker, clone 5TF1-1C2 est un anticorps dirigé contre le marqueur des maladies liées à une expansion de polyglutamine et dont l'utilisation a été validée en ELISA, IC, IH(P), IP et WB.
Sous-domaine de recherche
Maladies neurodégénératives
Test ELISA : 1/1 000-1/20 000
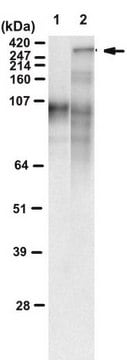
Analyse par western blotting : 1/1 000-1/20 000
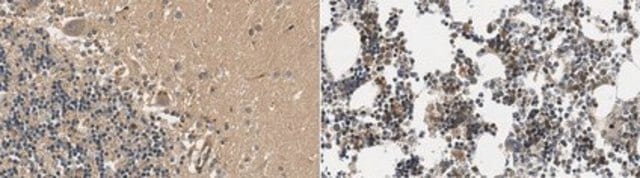
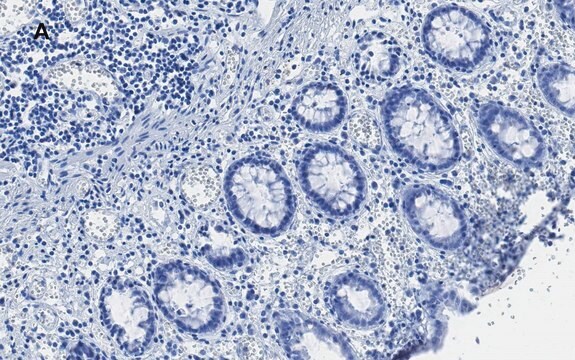
Immunohistochimie sur des coupes congelées ou incluses en paraffine (tissus humains) : 1/1 000-1/20 000
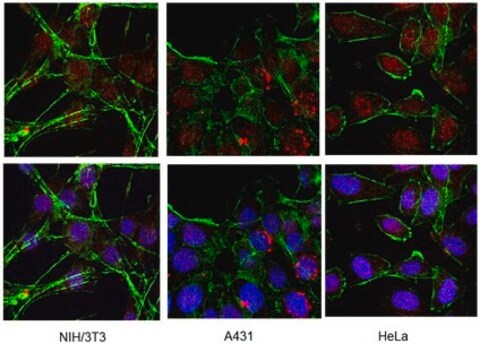
Immunocytochimie sur cellules transfectées : 1/1 000-1/20 000 Immunoprécipitation : 1/1 000-1/20 000
Il revient à l'utilisateur final de déterminer les dilutions de travail optimales.
Forme physique
Liquide d'ascite sans conservateurs.
Produit non purifié
Stockage et stabilité
À conserver à -20 °C pendant 1 an à compter de la date d'expédition. À diviser en aliquotes pour éviter les congélations et décongélations répétées. Pour récupérer le maximum de produit, centrifuger le flacon d'origine après décongélation et avant de retirer le capuchon.
Remarque sur l'analyse
Contrôle′Cerveau atteint de la chorée de Huntington
Autres remarques
Concentration : voir le certificat d'analyse du lot concerné.
Informations légales
CHEMICON is a registered trademark of Merck KGaA, Darmstadt, Germany
Clause de non-responsabilité
Sauf indication contraire dans notre catalogue ou toute autre documentation associée au(x) produit(s), nos produits sont uniquement destinés à la recherche et ne sauraient être utilisés à d'autres fins, ce qui inclut, sans s'y limiter, les utilisations commerciales non autorisées, les utilisations diagnostiques in vitro, les utilisations thérapeutiques ex vivo ou in vivo, ou tout type de consommation ou d'application chez l'être humain ou chez l'animal.