
05-583
Anti-CFTR Antibody, clone M3A7
clone M3A7, Upstate®, from mouse
Synonyme(s) :
ATP-binding cassette sub-family C, member 7, ATP-binding cassette transporter sub-family C member 7, cAMP-dependent chloride channel, cystic fibrosis transmembrane conductance regulator, cystic fibrosis transmembrane conductance regulator (ATP-binding ca
About This Item
Produits recommandés







Source biologique
mouse
Niveau de qualité
Forme d'anticorps
purified immunoglobulin
Type de produit anticorps
primary antibodies
Clone
M3A7, monoclonal
Espèces réactives
human
Fabricant/nom de marque
Upstate®
Technique(s)
immunohistochemistry: suitable
immunoprecipitation (IP): suitable
western blot: suitable
Isotype
IgG1
Numéro d'accès NCBI
Numéro d'accès UniProt
Conditions d'expédition
dry ice
Modification post-traductionnelle de la cible
unmodified
Informations sur le gène
human ... CFTR(1080)
Description générale
Spécificité
Immunogène
Application
This antibody has been reported to immunoprecipitate CFTR. (Kartner, N., 1998.)
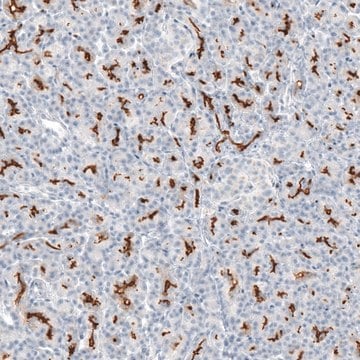
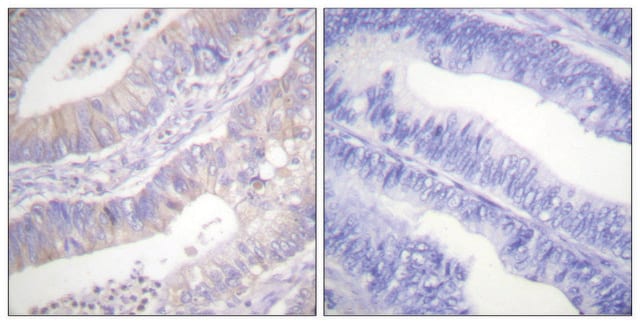
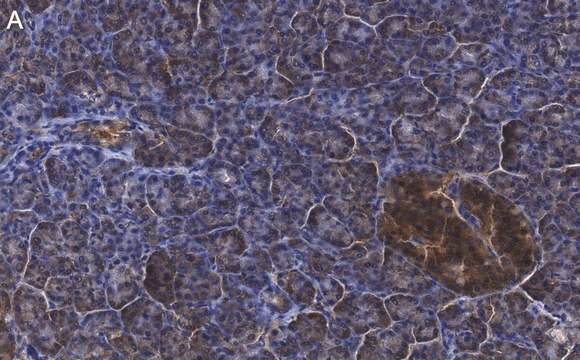
Immunohistochemistry:
This antibody has been reported to immunostain CFTR in human pancreatic tissue sections. (Kartner, N., 1998.)
Neuroscience
Ion Channels & Transporters
Qualité
Western Blot Analysis:
0.5-2 µg/mL of this lot detected CFTR from 20-50 µg of human T84 colon carcinoma epithelial RIPA cell lysates. 0.5-2 µg/mL of a previous lot detected CFTR from CFTR-transfected BHK (Haardt, M., 1999).
Note: Do not boil the lysate. Instead incubate at 37°C for 30 minutes. CFTR can run as a diffuse protein on SDS-PAGE.
Description de la cible
Forme physique
Stockage et stabilité
Handling Recommendations:
Upon receipt, and prior to removing the cap, centrifuge the vial and gently mix the solution. Aliquot into microcentrifuge tubes and store at -20°C. Avoid repeated freeze/thaw cycles, which may damage IgG and affect product performance. Note: Variability in freezer temperatures below -20°C may cause glycerol-containing solutions to become frozen during storage.
Remarque sur l'analyse
T84 cell lysate.
Autres remarques
Informations légales
Clause de non-responsabilité
Vous ne trouvez pas le bon produit ?
Essayez notre Outil de sélection de produits.
En option
Code de la classe de stockage
10 - Combustible liquids
Classe de danger pour l'eau (WGK)
WGK 1
Certificats d'analyse (COA)
Recherchez un Certificats d'analyse (COA) en saisissant le numéro de lot du produit. Les numéros de lot figurent sur l'étiquette du produit après les mots "Lot" ou "Batch".
Déjà en possession de ce produit ?
Retrouvez la documentation relative aux produits que vous avez récemment achetés dans la Bibliothèque de documents.
Notre équipe de scientifiques dispose d'une expérience dans tous les secteurs de la recherche, notamment en sciences de la vie, science des matériaux, synthèse chimique, chromatographie, analyse et dans de nombreux autres domaines..
Contacter notre Service technique