Allgemeine Beschreibung
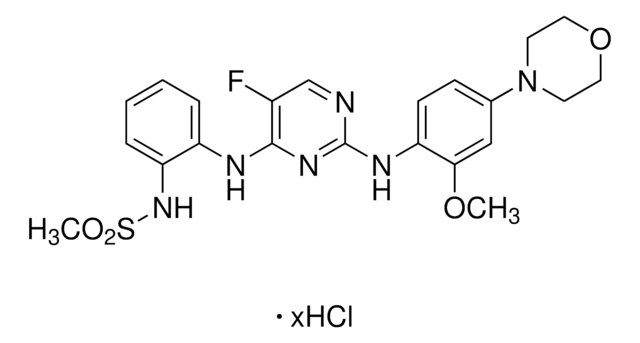
Ein zellpermeabler, ATP-kompetitiver, potenter und selektiver LRRK2-Inhibitor (IC50 von 13 nM, 6 nM und 2,45 µ;M für den Wildtyp, die G2019S-Mutante bzw. die arzneimittelresistente A2016T-Mutante LRRK2 in einem in vitro ATP-Site-kompetitiven Bindungsassay). Während es DCLK2 nachweislich hemmt (IC50 = 45 nM) und die MAPK7-Autophosphorylierung unterdrückt (EC50 = 160 nM), weist diese Verbindung (<;10 µ;M) eine sehr gute Gesamtselektivität auf, indem sie in einem Kinase-Bindungs- und biochemischen Test auf 12 von 442 anderen Kinasen abzielt. Bei 0,05–;3 µ;M induziert es eine dosisabhängige Hemmung der Phosphorylierung von Ser910 und Ser935, begleitet von einem Verlust der 14-3-3-Bindung sowohl an Wildtyp-LRRK2 als auch an LRRK2[G2019S] in stabil transfizierten HEK293-Zellen, jedoch nicht in den arzneimittelresistenten LRRK2[A2016T] und LRRK2[A2016T + G2019S]-Mutanten. Ähnliche Auswirkungen auf die endogene LRRK2-Phosphorylierung und die 14-3-3-Bindung können in menschlichen lympho¬;blastoiden Zellen und für die LRRK2-Mutante G2019S, die von einem Parkinson-Patienten stammt, sowie in menschlichen Neuroblastom-Zellen SHSY5Y und in Schweizer 3T3-Zellen der Maus beobachtet werden. 100 mg/kg des Wirkstoffs, der Mäusen injiziert wird, bewirkt eine vollständige Ser910- und Ser935-Dephosphorylierung von LRRK2 in der Niere, aber nicht im Gehirn, was darauf zurückzuführen sein könnte, dass die Blut-Hirn-Schranke nicht durchdrungen werden kann. Darüber hinaus fördert es die Relokalisierung von LRRK2 zu mehr aggregierten und fibrillenartigen Strukturen.
Ein zellpermeabler, ATP-kompetitiver, potenter und selektiver LRRK2-Inhibitor (IC50 von 13 nM, 6 nM und 2,45 µ;M für den Wildtyp, die G2019S-Mutante bzw. die arzneimittelresistente A2016T-Mutante LRRK2 in einem in vitro ATP-Site-kompetitiven Bindungsassay). Während es DCLK2 nachweislich hemmt (IC50 = 45 nM) und die MAPK7-Autophosphorylierung unterdrückt (EC50 = 160 nM), weist diese Verbindung (<;10 µ;M) eine sehr gute Gesamtselektivität auf, indem sie in einem Kinase-Bindungs- und biochemischen Test auf 12 von 442 anderen Kinasen abzielt. Bei 0,05-3 µ;M induziert es eine dosisabhängige Hemmung der Phosphorylierung von Ser910 und Ser935, begleitet von einem Verlust der 14-3-3-Bindung sowohl an Wildtyp-LRRK2 als auch an LRRK2[G2019S] in stabil transfizierten HEK293-Zellen, jedoch nicht in den arzneimittelresistenten LRRK2[A2016T] und LRRK2[A2016T + G2019S]-Mutanten. ′;Ähnliche Auswirkungen auf die endogene LRRK2-Phosphorylierung und die 14-3-3-Bindung können in menschlichen lympho-blastoiden Zellen und für die LRRK2-Mutante G2019S, die von einem Parkinson-Patienten stammt, sowie in menschlichen Neuroblastom-Zellen SHSY5Y und in Schweizer 3T3-Zellen der Maus beobachtet werden. 100 mg/kg des Wirkstoffs, der Mäusen injiziert wird, bewirkt eine vollständige Ser910- und Ser935-Dephosphorylierung von LRRK2 in der Niere, aber nicht im Gehirn, was darauf zurückzuführen sein könnte, dass die Blut-Hirn-Schranke nicht durchdrungen werden kann. Darüber hinaus fördert es die Relokalisierung von LRRK2 zu mehr aggregierten und fibrillenartigen Strukturen.
Sonstige Hinweise
Deng, X., et al. 2011. Nat. Chem. Biol.7, 203.