
WH0002548M1
Monoclonal Anti-GAA antibody produced in mouse
clone 3C6, purified immunoglobulin, buffered aqueous solution
Synonyme(s) :
Anti-LYAG, Anti-glucosidase, α acid (Pompe disease, glycogen storage disease type II)
About This Item
Produits recommandés
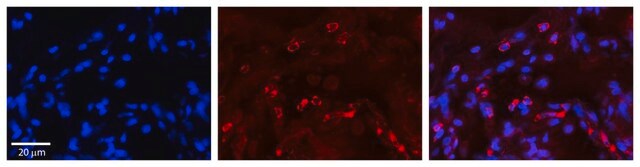


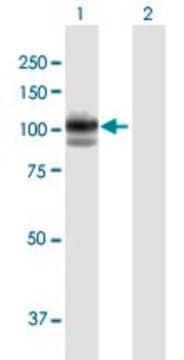

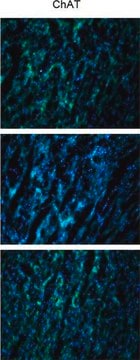
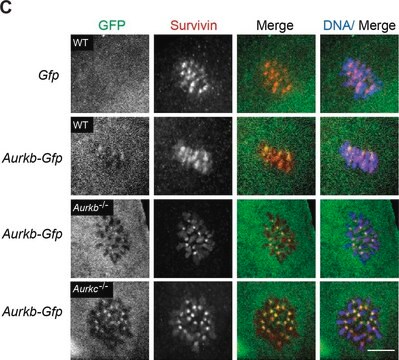


Source biologique
mouse
Conjugué
unconjugated
Forme d'anticorps
purified immunoglobulin
Type de produit anticorps
primary antibodies
Clone
3C6, monoclonal
Forme
buffered aqueous solution
Espèces réactives
human
Technique(s)
indirect ELISA: suitable
western blot: 1-5 μg/mL
Isotype
IgG1κ
Numéro d'accès GenBank
Numéro d'accès UniProt
Conditions d'expédition
dry ice
Température de stockage
−20°C
Modification post-traductionnelle de la cible
unmodified
Informations sur le gène
human ... GAA(2548)
Description générale
Immunogène
Sequence
GEARGELFWDDGESLEVLERGAYTQVIFLARNNTIVNELVRVTSEGAGLQLQKVTVLGVATAPQQVLSNGVPVSNFTYSPDTKVLDICVSLLMGEQFLVSWC
Actions biochimiques/physiologiques
Forme physique
Informations légales
Clause de non-responsabilité
Not finding the right product?
Try our Outil de sélection de produits.
Code de la classe de stockage
10 - Combustible liquids
Point d'éclair (°F)
Not applicable
Point d'éclair (°C)
Not applicable
Équipement de protection individuelle
Eyeshields, Gloves, multi-purpose combination respirator cartridge (US)
Certificats d'analyse (COA)
Recherchez un Certificats d'analyse (COA) en saisissant le numéro de lot du produit. Les numéros de lot figurent sur l'étiquette du produit après les mots "Lot" ou "Batch".
Déjà en possession de ce produit ?
Retrouvez la documentation relative aux produits que vous avez récemment achetés dans la Bibliothèque de documents.
Notre équipe de scientifiques dispose d'une expérience dans tous les secteurs de la recherche, notamment en sciences de la vie, science des matériaux, synthèse chimique, chromatographie, analyse et dans de nombreux autres domaines..
Contacter notre Service technique