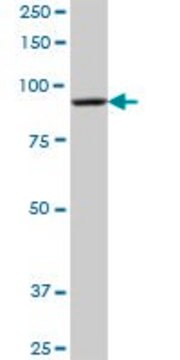
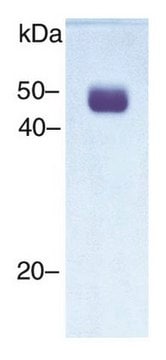
Synthetic peptide directed towards the C terminal region of human GCDH
Biochem/physiol Actions
GCDH belongs to the acyl-CoA dehydrogenase family. It catalyzes the oxidative decarboxylation of glutaryl-CoA to crotonyl-CoA and CO(2) in the degradative pathway of L-lysine, L-hydroxylysine, and L-tryptophan metabolism. It uses electron transfer flavoprotein as its electron acceptor. The enzyme exists in the mitochondrial matrix as a homotetramer of 45-kD subunits.The protein encoded by this gene belongs to the acyl-CoA dehydrogenase family. It catalyzes the oxidative decarboxylation of glutaryl-CoA to crotonyl-CoA and CO(2) in the degradative pathway of L-lysine, L-hydroxylysine, and L-tryptophan metabolism. It uses electron transfer flavoprotein as its electron acceptor. The enzyme exists in the mitochondrial matrix as a homotetramer of 45-kD subunits. Alternatively spliced transcript variants encoding different isoforms have been identified.
Sequence
Synthetic peptide located within the following region: IARQARDMLGGNGISDEYHVIRHAMNLEAVNTYEGTHDIHALILGRAITG
Physical form
Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and 2% sucrose.
Disclaimer
Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to humans or animals.
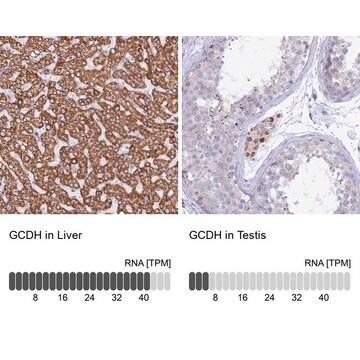
Glutaryl-CoA dehydrogenase (GCDH) is a mitochondrial enzyme that is involved in the degradation of tryptophan, lysine and hydroxylysine. Deficient enzyme activity leads to glutaric aciduria type-I (GA-I). This neurometabolic disease usually manifests with acute encephalopathic crises and striatal neuronal death
Glutaric aciduria type 1 (GA1) is a rare inherited metabolic disorder caused by a deficiency of glutaryl-coenzyme A dehydrogenase (GCDH), with accumulation of neurotoxic metabolites, resulting in a complex movement disorder, irreversible brain damage, and premature death in untreated individuals.
Glutaric aciduria type I (GA-1) is an inborn error of metabolism with a severe neurological phenotype caused by the deficiency of glutaryl-coenzyme A dehydrogenase (GCDH), the last enzyme of lysine catabolism. Current literature suggests that toxic catabolites in the brain
Questions
Reviews
★★★★★ No rating value
Active Filters
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.