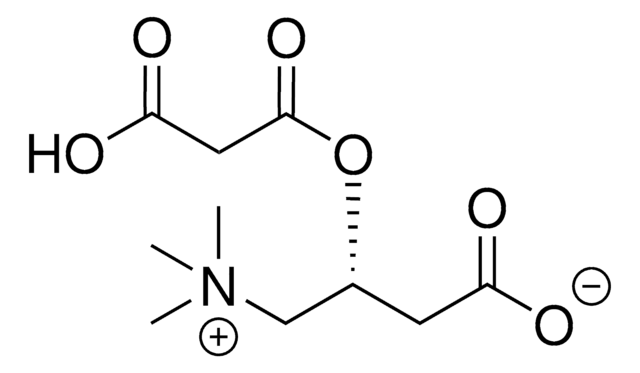
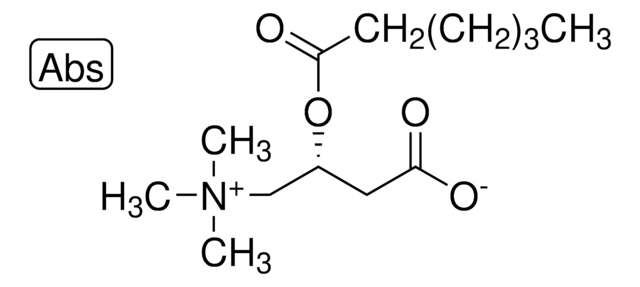
Increased formation and excretion of glutarylcarnitin results from a glutaryl-CoA dehydrogenase deficiency, an inborn error of lysine and tryptophan metabolism. Secondary carnitine depletion due to increased formation and urinary excretion of glutarylcarnitine is suggested to play an important role in the neuropathogenesis of glutaryl-CoA dehydrogenase deficiency, inducing excitotoxic neuronal damage and mitochondrial dysfunction.
Clinica chimica acta; international journal of clinical chemistry, 164(3), 261-266 (1987-05-15)
A technique for the identification of glutarylcarnitine in urine from a patient with glutaric aciduria type 1 is described. The patient's urine sample was partially purified using an anion exchange column and analyzed by a carboxylic acid analyzer fitted with
Glutaryl-CoA dehydrogenase deficiency is an inherited organic aciduria with predominantly neurological presentation. Biochemically, it is characterized by an accumulation and enhanced urinary excretion of two key organic acids, glutaric acid and 3-hydroxyglutaric acid. If untreated, acute striatal degeneration is often
Glutaric aciduria type I (GA-I) is an inborn error of lysine and tryptophan metabolism. Clinical manifestations of GA-I include dystonic or dyskinetic cerebral palsy, but when the symptoms occur, treatment is not effective. In Taiwan, newborn screening for GA-I started
Glutaric aciduria; a "new" disorder of amino acid metabolism.
To determine whether circulating metabolic intermediates are related to insulin resistance and beta-cell dysfunction in individuals at risk for type 2 diabetes. In 73 sedentary, overweight to obese, dyslipidemic individuals, insulin action was derived from a frequently sampled intravenous glucose
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.