
SAB2108553
Anti-FAH
IgG fraction of antiserum
About This Item
Productos recomendados



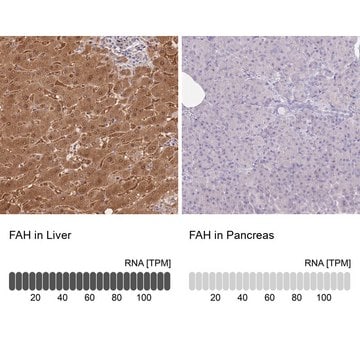

biological source
rabbit
Quality Level
conjugate
unconjugated
antibody form
IgG fraction of antiserum
antibody product type
primary antibodies
clone
polyclonal
form
buffered aqueous solution
mol wt
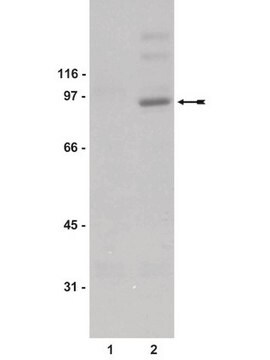
46 kDa
species reactivity
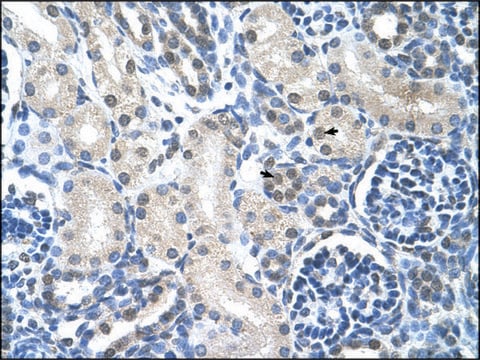
dog, horse, human, mouse, rat
concentration
0.5-1 mg/mL
technique(s)
immunoblotting: suitable
immunohistochemistry: suitable
accession no.
NM_000137
UniProt accession no.
shipped in
wet ice
storage temp.
−20°C
target post-translational modification
unmodified
Gene Information
human ... FAH(2184)
General description
Immunogen
Biochem/physiol Actions
Sequence
Physical form
Disclaimer
¿No encuentra el producto adecuado?
Pruebe nuestro Herramienta de selección de productos.
Storage Class
10 - Combustible liquids
wgk_germany
WGK 3
flash_point_f
Not applicable
flash_point_c
Not applicable
Certificados de análisis (COA)
Busque Certificados de análisis (COA) introduciendo el número de lote del producto. Los números de lote se encuentran en la etiqueta del producto después de las palabras «Lot» o «Batch»
¿Ya tiene este producto?
Encuentre la documentación para los productos que ha comprado recientemente en la Biblioteca de documentos.
Nuestro equipo de científicos tiene experiencia en todas las áreas de investigación: Ciencias de la vida, Ciencia de los materiales, Síntesis química, Cromatografía, Analítica y muchas otras.
Póngase en contacto con el Servicio técnico