
B8299
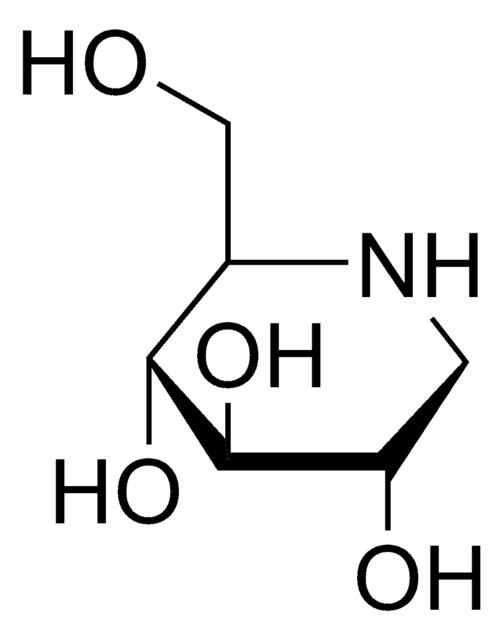
N-Butyldeoxynojirimycin
film (dried in situ), ≥98% (TLC)
Synonym(s):
Miglustat, NB-DNJ
About This Item
Recommended Products







product name
N-Butyldeoxynojirimycin, film (dried in situ)
assay
≥98% (TLC)
Quality Level
form
film (dried in situ)
solubility
water: 9.80-10.20 mg/mL, clear, colorless
storage temp.
2-8°C
SMILES string
CCCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO
InChI
1S/C10H21NO4/c1-2-3-4-11-5-8(13)10(15)9(14)7(11)6-12/h7-10,12-15H,2-6H2,1H3/t7-,8+,9-,10-/m1/s1
InChI key
UQRORFVVSGFNRO-UTINFBMNSA-N
Gene Information
human ... UGCG(7357)
Related Categories
General description
Application
- in the inhibition of glycolipid synthesis in neuroblastoma cells
- in the inhibition the ceramide-specific glycosyltransferase in hepatocytes
- in the inhibition of β-glucosidase (GBA2) using fluorescence- activity assay in human embryonic kidney (HEK293) cells.
Biochem/physiol Actions
Storage Class
11 - Combustible Solids
wgk_germany
WGK 3
flash_point_f
Not applicable
flash_point_c
Not applicable
ppe
Eyeshields, Gloves, type N95 (US)
Certificates of Analysis (COA)
Search for Certificates of Analysis (COA) by entering the products Lot/Batch Number. Lot and Batch Numbers can be found on a product’s label following the words ‘Lot’ or ‘Batch’.
Already Own This Product?
Find documentation for the products that you have recently purchased in the Document Library.
Customers Also Viewed





Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.
Contact Technical Service