
SAB2100872
Anti-GAA antibody produced in rabbit
affinity isolated antibody
Sinónimos:
Anti-Glucosidase, α; acid, Anti-LYAG
About This Item
Productos recomendados








origen biológico
rabbit
Nivel de calidad
conjugado
unconjugated
forma del anticuerpo
affinity isolated antibody
tipo de anticuerpo
primary antibodies
clon
polyclonal
Formulario
buffered aqueous solution
mol peso
98 kDa
reactividad de especies
dog, guinea pig, human, rat, mouse, bovine
concentración
0.5 mg - 1 mg/mL
técnicas
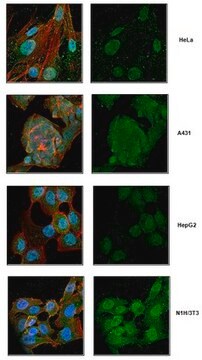
immunohistochemistry: suitable
western blot: suitable
Nº de acceso UniProt
Condiciones de envío
wet ice
temp. de almacenamiento
−20°C
modificación del objetivo postraduccional
unmodified
Información sobre el gen
human ... GAA(2548)
Descripción general
Inmunógeno
Aplicación
Acciones bioquímicas o fisiológicas
Secuencia
Forma física
Cláusula de descargo de responsabilidad
¿No encuentra el producto adecuado?
Pruebe nuestro Herramienta de selección de productos.
Código de clase de almacenamiento
10 - Combustible liquids
Clase de riesgo para el agua (WGK)
WGK 3
Punto de inflamabilidad (°F)
Not applicable
Punto de inflamabilidad (°C)
Not applicable
Elija entre una de las versiones más recientes:
Certificados de análisis (COA)
¿No ve la versión correcta?
Si necesita una versión concreta, puede buscar un certificado específico por el número de lote.
¿Ya tiene este producto?
Encuentre la documentación para los productos que ha comprado recientemente en la Biblioteca de documentos.
Nuestro equipo de científicos tiene experiencia en todas las áreas de investigación: Ciencias de la vida, Ciencia de los materiales, Síntesis química, Cromatografía, Analítica y muchas otras.
Póngase en contacto con el Servicio técnico