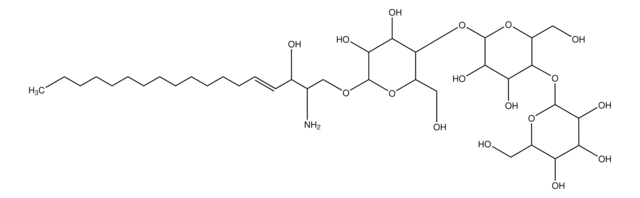
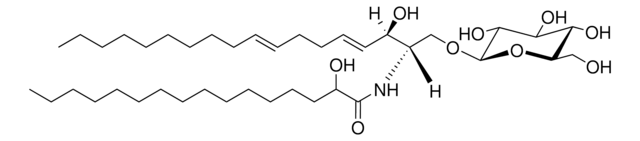
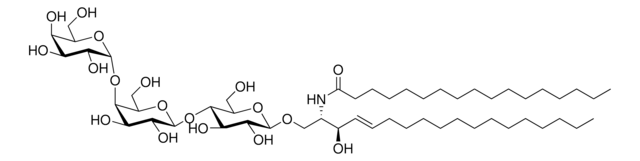
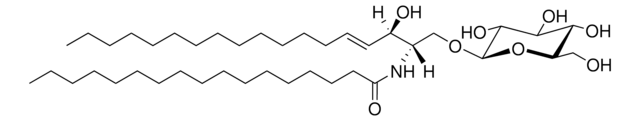
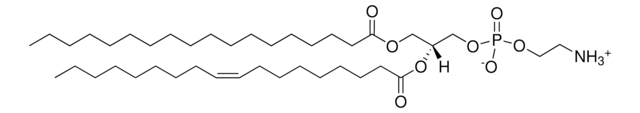
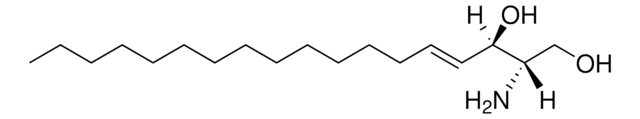
Glucosylsphingosine is a cytotoxic compound. Accumulation of glucosylsphingosine in brain and other tissues occurs in patients with Gaucher disease, which is an inherited deficiency of lysosomal glucocerebrosidase, which converts glucosylsphingosine to glucose and sphingosine.
Gaucher disease (GD) is caused by mutations on the GBA1 gene leading to deficiency in acid β-glucosidase (GCase) and subsequent accumulation of its substrates, glucosylceramide (GlcC) and glucosylsphingosine (GlcS). GlcS in plasma has been proposed as a highly sensitive and
Gaucher disease (GD) is caused by a deficiency of β-glucosidase (GCase), leading to accumulation of glucosylceramide (GlcC) and glucosylsphingosine (Lyso-Gb1). Lyso-Gb1 is a reliable biomarker for GD. This study aims to develop a simple, effective and accurate method for the
Splenectomy in children with the Norrbottnian type of Gaucher disease is followed by increased blood levels of glucosylceramide and impaired neurological and mental status. High blood levels are associated with an increased accumulation of glucosylceramide in perivascular Gaucher cells in
Journal of internal medicine, 246(6), 587-590 (2000-01-05)
Chronic Gaucher disease [GD] in association with systemic AL amyloidosis is extremely rare. We describe a 46-year-old Greek male with chronic GD confirmed by low glucocerebroside activity in fibroblasts and N370S/L444P mutations at the cerebrosidase gene, who also had systemic
Gaucher disease is a glycolipid storage disorder characterized by accumulation of glucocerebroside in the liver, spleen, and bones, and caused by a deficiency of glucocerebrosidase. Glucocerebrosidase cDNA has been cloned and sequenced, and much has been learned about the synthesis
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.