
推荐产品








品質等級
化驗
≥98.0% (TLC)
形狀
powder
脂質類型
sphingolipids
儲存溫度
−20°C
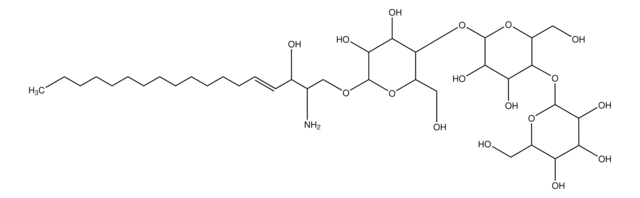
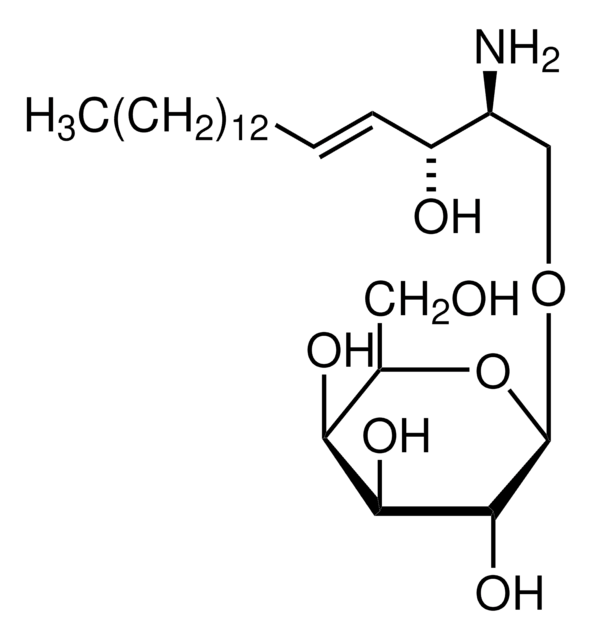
SMILES 字串
O[C@@H]1[C@@H](O)[C@H](OC[C@H](N)[C@H](O)/C=C/CCCCCCCCCCCCC)O[C@H](CO)[C@H]1O
InChI
1S/C24H47NO7/c1-2-3-4-5-6-7-8-9-10-11-12-13-14-15-19(27)18(25)17-31-24-23(30)22(29)21(28)20(16-26)32-24/h14-15,18-24,26-30H,2-13,16-17,25H2,1H3/b15-14+/t18-,19+,20?,21+,22+,23?,24+/m0/s1
InChI 密鑰
HHJTWTPUPVQKNA-JLRUQHRASA-N
生化/生理作用
Glucosylsphingosine is a cytotoxic compound. Accumulation of glucosylsphingosine in brain and other tissues occurs in patients with Gaucher disease, which is an inherited deficiency of lysosomal glucocerebrosidase, which converts glucosylsphingosine to glucose and sphingosine.
儲存類別代碼
11 - Combustible Solids
水污染物質分類(WGK)
WGK 3
閃點(°F)
Not applicable
閃點(°C)
Not applicable
N G Conradi et al.
Acta neuropathologica, 75(4), 385-390 (1988-01-01)
Splenectomy in children with the Norrbottnian type of Gaucher disease is followed by increased blood levels of glucosylceramide and impaired neurological and mental status. High blood levels are associated with an increased accumulation of glucosylceramide in perivascular Gaucher cells in
Lulu Kang et al.
Journal of human genetics, 62(8), 763-768 (2017-03-31)
Gaucher disease (GD) is an inherited metabolic disorder that involves accumulation of glycolipid glucocerebroside in monocyte-macrophage cells, which can result in multiple organ damage. Enzyme replacement and substrate reduction therapies have improved the potential for early diagnosis and treatment. Determining
A Kaloterakis et al.
Journal of internal medicine, 246(6), 587-590 (2000-01-05)
Chronic Gaucher disease [GD] in association with systemic AL amyloidosis is extremely rare. We describe a 46-year-old Greek male with chronic GD confirmed by low glucocerebroside activity in fibroblasts and N370S/L444P mutations at the cerebrosidase gene, who also had systemic
E Beutler
Blood reviews, 2(1), 59-70 (1988-03-01)
Gaucher disease is a glycolipid storage disorder characterized by accumulation of glucocerebroside in the liver, spleen, and bones, and caused by a deficiency of glucocerebrosidase. Glucocerebrosidase cDNA has been cloned and sequenced, and much has been learned about the synthesis
A convenient approach to facilitate monitoring Gaucher disease progression and therapeutic response.
Wujuan Zhang et al.
The Analyst, 142(18), 3380-3387 (2017-08-16)
Gaucher disease (GD) is caused by mutations on the GBA1 gene leading to deficiency in acid β-glucosidase (GCase) and subsequent accumulation of its substrates, glucosylceramide (GlcC) and glucosylsphingosine (GlcS). GlcS in plasma has been proposed as a highly sensitive and
我们的科学家团队拥有各种研究领域经验,包括生命科学、材料科学、化学合成、色谱、分析及许多其他领域.
联系技术服务部门