
1 財団法人九州先端科学技術研究所
2 九州大学 未来化学創造センター 客員准教授
3 九州大学 未来化学創造センター 教授
材料科学の基礎 第1号 2009→ PDF版
Article outline
§1.有機EL
§1-1.はじめに
§1-2.有機ELの開発の歴史 -1-
§1-3.有機ELの開発の歴史 -2-
§2.有機ELの動作機構および構造
§2-1.有機ELの素過程
§2-2.有機EL素子構造
§3.有機ELの作製方法
§3-1.材料の昇華精製
§3-2.ITO基板の取り扱い
§3-3.真空蒸着装置を用いた有機ELの作製
§3-4.特性評価
§1.有機EL
§1-1.はじめに
「有機発光材料」とは、ある種のエネルギー刺激を与えられたとき、それに対する応答として光を放出する機能を有する有機材料を言う。有機材料の発光過程は一種のエネルギー変換プロセスとしてとらえることが出来る。刺激源である入力エネルギー種としては、機械エネルギー、光エネルギー、電気エネルギー、化学エネルギー等多種多様である。それぞれの刺激に対応してメカノルミネッセンス(Mechanoluminescence)、フォトルミネッセンス(Photoluminescence)、エレクトロルミネッセンス(Electroluminescence、EL)、ケミルミネッセンス(Chemiluminescence)と呼ばれている。各発光現象の詳細なプロセスには異なるところがあるが、共通している点はどんな発光現象でも有機分子が最終的に必ず高いエネルギーを持つ励起状態を形成し、そのエネルギーを光として放出し、エネルギー的に低く安定な基底状態に戻るプロセスを含んでいる点である。その中でも、エレクトロルミネッセンスとは、蛍光体に電気エネルギーを与えて励起させ、励起状態から失活する際のエネルギーを光として取り出す現象をいう。このエレクトロルミネッセンス現象を利用した発光素子に有機ELがある(図1-1)。有機ELは有機薄膜内にキャリアを注入し、蛍光色素上で再結合させて励起状態を形成し、発光を取り出すことからキャリア注入型ELと呼ばれる。日本では、有機ELという呼び名が定着したが、世界的には、発光ダイオードの一種として、OLED(Organic Light Emitting Diode)という呼び名が一般的な表現である。

図1-1 有機EL素子の典型的な構造と発光の様子
また、別の切り口から有機ELを見ると、「有機半導体」というキーワードが浮かび上がる。有機物は、シリコン半導体に牽引されるエレクトロニクスの分野において、プラスチックやゴム、紙などから連想されるように、古くからその絶縁特性や誘電体としての特性が利用され、絶縁被覆材料、コンデンサー、筐体などの構造形成物としてしか捉えられてこなかった。しかし、有機ELや有機トランジスタなど有機電子デバイスの研究開発が進展し、動作原理が理解されるにつれ、導電率から見ると絶縁体として分類される場合でも、ある程度の移動度で電子とホールが移動し電流が観測される有機材料が多数見つけられ、有機半導体として呼ばれるようになった。有機ELでは、mA/cm2オーダーの電流密度を制御できるようになり、昨今の研究の進捗により、物性値としての導電率は未だに低いものの、材料開発や素子構造の最適化によりkA/cm2もの電流密度を制御できる時代に突入している。
有機EL素子は、
- 電流注入型薄膜面発光デバイスであり、視野角が広く視認性に優れる。
- 低電圧駆動が可能である。
- 応答速度が速く、動画再生性能に優れる。
- 単純な素子構造のため、薄型化および軽量化できる。
などの特長を有しており、高機能フラットパネルへの応用の期待から、活発に研究開発が行われているデバイスの一つである。
§1-2.有機ELの開発の歴史 -1-
その有機ELの研究は、1953年に有機色素を含む高分子薄膜に、高い交流電流を印加すると発光することを発見したA.Bernanoseの研究が始まりと言われている。彼らは、この色素含有高分子薄膜からの発光が、既に知られていたキャリア注入を伴わない真性ELの一種である無機ELと同様の機構で起こると主張した1が、この有機物からの発光は、放電に由来する紫外光によって蛍光体が励起されたことによる二次的発光であったと現在では理解されている。さらに、1960年代に入ってNew York大のM. PopeらやNRC CanadaのW. Helfrichらがアントラセン(図1-2)単結晶の両端に、アントラセンのアニオンラジカルとカチオンラジカルを含む溶液をそれぞれカソード、アノードとして用いて、電場を印加すると、単結晶からアントラセンのフォトルミネッセンスと同じ蛍光が得られることを示した2-4。液体電極を介して有機薄膜にキャリアを注入し、アントラセンからの発光を取り出したこのW. Helfrich等の研究が、本当の意味での有機ELの研究の始まりといえる。

図1-2 アントラセン
このように、有機ELはこれまで絶縁性と考えられていた有機物に電界を印加することによって注入された正と負のキャリアが、有機分子上で再結合して励起子を生成し、その輻射失活によって発光するという非常に興味深い現象を利用したデバイスである。そのため、様々な方法で高輝度・高効率化が図られたが、1970年代から1980年代前半は、有機薄膜を用いたキャリア注入型ELの模索が続いた。
第一の課題は、キャリアの注入、特に電子の注入であった。コピー機やレーザープリンタの感光ドラムに利用される有機光導電体(Organic Photoconductor:OPC)材料として、ポリビニルカルバゾール5やトリフェニルアミン誘導体6,7のホール輸送特性が見いだされて以来、ホールの注入および輸送が起こる有機材料は数多く報告されていたが、その有機材料の持つホールとの親和性のために電子の注入は起こりにくく、電子注入に有利な有機材料はほとんどなかった。さらに、有機物に電子を注入するために仕事関数の小さなアルカリ金属やアルカリ土類金属の固体電極が用いられたが、金属の活性が高く、空気中で安定して用いることができなかった。
第二の課題は、電界印加時の有機薄膜の安定性不足であった。様々な有機色素の真空蒸着膜が有機ELに試みられたが、蒸着膜は目的とした単結晶とはならず微結晶集合体であり、電界印加時の絶縁破壊や放電現象が不安定性の原因であった。そこで、印加電圧を下げる目的でラングミュアーブロジット(LB)膜を用いた超薄膜有機ELが試みられたが、思うような安定性は得られなかった8,9。1980年半ばには、S. Hayashi等は、蒸着ペリレン薄膜を用いた素子においてホール注入を改良するため、インジウム- スズ酸化物(Indium-Tin Oxide:ITO)透明電極と発光層であるペリレン(図1-3)層の間にポリチオフェン薄膜を導入した積層型素子を作製し、著しいホール注入特性の改善と発光開始電圧の大幅な低下を報告した10。しかしながら、ポリチオフェン膜の導電率が高くなかったため、当時はポリチオフェンをホール注入電極と見なし、絶縁性のホール注入・輸送層とは考えなかったため、機能分離した多層構造の有機ELの発想には至らなかった。

図1-3 ペリレン
§1-3.有機ELの開発の歴史 -2-
コダック社C. W. Tangによる高効率有機ELの実現からリン光発光有機ELへ
この有機ELの研究の滞った状態を破ったのは、1987年にC.W. Tang等によって発表された100 nmオーダーの有機超薄膜の積層構造を採用した有機EL素子である11。彼らは、ITOガラス基板上にホール輸送層であるジアミン誘導体75 nmと電子輸送層兼発光層であるTris(8-quinolinolate)aluminum(Alq3)60 nm、MgAg電極を順次真空蒸着して素子を作製した。この素子にITO電極に対して順方向の電圧を印加すると、10 V以下の低電圧で1,000 cd/m2を越えて発光し、外部量子効率1%を超える高効率を示し、当時の有機ELとしては考えられない高性能の有機EL素子を実現した。そのデバイス構造を図1-4に示す。

図1-4 C. W. Tang等が発表した有機ELの構造と有機材料
この有機ELの特徴は、
- 電子的・光学的性質が異なる有機薄膜を二層組み合わせたこと。
- 二層それぞれの厚さがそれまで考えられていた有機層よりも1桁程度薄いこと。
- 電子注入には有利だが大気中で不安定だったマグネシウム電極に銀を少量混ぜ合金にすることで、電子注入特性を犠牲にせずに大気安定性を得たことと、有機物との良好な密着性を達成したこと。
であり、彼らの積層型有機EL素子の成功は様々な工夫の上に成り立っていた。このC. W. Tang等の報告は、有機ELのブレイクスルーと呼ばれるほど大きな影響を与え、この高輝度・高効率の有機ELの発表によって有機ELの研究は一気に加速された。その後、C. Adachi等は発光層を電子輸送層とホール輸送層で挟んだ三層型構造を試み(図1-5)、さらにホール輸送層が発光層の機能をかねる新しい二層型構造の利用が可能であることを実験的に示し、C. W. Tang等の二層構造が必ずしも唯一可能な積層構造でないことを示した12,13。

図1-5 最初の有機ダブルへテロ構造12
1990年代に入ると、さらに世界中の研究者によって有機ELの研究が活発に行われるようになった。C. Hosokawa等は新規材料を分子設計する立場から研究を展開し、ジスチリルアリーレン誘導体(図1-6)という一群の高性能の青色発光材料を発表した14。Y. Hamada等はAlq3蒸着膜が優れた耐久性を持つことを重視して、それを出発点として新しい発光性の金属錯体を広範囲に探索し、いくつかの優れたEL材料を発見した15。また、銅フタロシアニンやスターバーストポリアミンをITO電極とホール輸送層(TPD)の間に挟むことで耐久性を著しく向上させるバッファ層の考え方が提案されその研究も進んだ16。この時期に報告されたホール輸送材料であるトリフェニルアミン誘導体(TPD)12、電子輸送材料であるオキサジアゾール誘導体(PBD)13、発光材料(ホスト材料)であるAlq311、青色発光材料であるジスチリルアリレン誘導体14が基本骨格として進化し、現在の有機ELに用いられ、材料開発の基礎になっている。

図1-6 4,4'-bis(2,2-diphenylethenyl)biphenyl
これまで述べてきたキレート金属錯体などの低分子色素を用いた素子の発展と同様に、π共役高分子材料もポリフェニレンビニレン(PPV)の単層薄膜で、キャリア注入型ELが観察された17のをきっかけに、ポリアリキルチオフェン(PAT)18、ポリアルキルフルオレン(PF)19などが用いられるようになった(図1-7)。また、π共役高分子を用いたELにおいても電子輸送性のオキサジアゾール誘導体を分散したポリメタクリル酸メチルを積層させて、発光効率を向上させる試みも行われた20。

図1-7 PATとPF
さらに、有機材料の探索とともに、赤(スペクトルのピーク波長約625 nm)、緑(約520 nm)、青(約460 nm)に発光スペクトルを持つ色素を組み合わせることによって白色ELの実現21や、陰極電極にフッ化リチウムを挿入することによって、著しく性能を向上させた研究などがあった22。さらに、有機ELを単なる発光素子としてではなく、素子にファブリペロー型ミラーを導入した微小共振器ELの作製23,24や、電流励起による有機導波路型レーザーダイオードの実現に向けた厚膜素子の開発も研究された25。
最近の特筆すべき研究に、遷移重金属を用いたリン光性の発光材料の開発がある。Princeton大とSouthern California大のグループから、白金錯体やイリジウム錯体をドーパントに用いた有機ELから、室温で安定したリン光発光が取り出せることが報告され26,27、リン光有機ELの研究開発が一気に進んだ28。これまでにも、九州大学29やNTT30のグループによって、ベンゾフェノン誘導体やケトクマリンを用い、リン光発光の有機ELが確認されていたが、77 Kの低温下に限られていた。彼らが作製した三重項励起子からの発光を利用するデバイスは、有機ELの一重項励起子からの発光を利用した理論上の最大量子効率である5%を越える8%を達成した。現在では、イリジウム錯体の積極的な開発や、ホスト材料の三重項励起エネルギー状態も徐々に明らかにされ、C. AdachiやS. Tokito等により最適な素子構造を用いることによって、緑 ≒ 19% 31、赤 ≒ ~ 12% 32、青 ≒ 6 ~20% 33,34の非常に高い外部量子効率が実現され、特に、緑に至ってはほぼ理論限界の発光効率まで達している。
そして、ついに1990年代終わりには有機ELの実用化が始まり、1987年のブレイクスルーから約10年間で有機ELは大きく発展し、さらに、2007年12月にソニーから発売された有機ELテレビによって実を結んだといえる(図1-8)。現在、有機半導体デバイスはグリーンエレクトロニクスとして、環境負荷が小さく、高効率な電子デバイスの実現が期待され脚光を浴びている。さらに、有機物ならではの特色として、印刷法によって電子デバイスが作製できることや、フレキシブル性(図1-9)、つまり、プラスチック製の下敷きのように軽く、落としても割れない性質も着目され、有機半導体デバイスの研究開発はさらに盛んになっている。

図1-8 ソニーより発売された有機ELテレビXL-1

図1-9 試作したフレキシブル有機EL
§2.有機ELの動作機構および構造
§2-1.有機ELの素過程
まず、有機ELを作製/評価するために理解することが必要な有機ELの動作機構や効率について、有機EL素子の動作機構を素過程(図2-1)を用いて説明する。(効率に関する詳細な説明は「Appendix Ⅰ(PDF版)」を参照)
- 外部から電場が印加され、陽極からホールがホール輸送層へ注入され、陰極から電子が電子輸送層に注入される。(キャリアの注入バランス:γ)
- 注入されたキャリアが、分子間をホッピング移動する。
- 発光層でホールと電子は再結合し、電気的に中性な励起子を生成する。(発光性(一重項)励起子の生成効率:ηr)
- 励起子は、蛍光量子効率(三重項励起子からの発光も考慮する場合は、発光の量子効率)に従って光を発して輻射失活する。(蛍光量子効率:ηf)
- 有機層中で発生した光が、光取り出し面から空気中へ取り出される。(光取り出し効率:ηext)

図2-1 有機ELのキャリア注入から発光にいたるまでの素過程
キャリア注入過程
キャリア注入型デバイスである有機EL素子では、外部から印加した電圧によって、電極からキャリアが注入される。次節で詳しく説明するが、下記の素子構造を考えてみる。(図2-2参照)
ITO / α-NPD(50 nm)/ Alq3(50 nm)/ MgAg(150 nm)/ Ag(10 nm)
この時、例えばα-NPDの-5.5 eVの最高占有軌道(Highest Occupied Molecular Orbital:HOMO)にITO電極からホールを注入するため、ITO電極の仕事関数はα-NPDのHOMOにマッチングするよう大きい方が良い。溶媒を用いて超音波洗浄したITO表面の仕事関数は、およそ-4.7 eV程度であるが、UV-オゾン洗浄することによって、溶媒を用いた洗浄で取り残した有機物の除去とITOの表面酸化がなされ仕事関数は-5.0 eV程度まで上昇することが、紫外光電子分光法(Ultraviolet Photoelectron Spectroscopy:UPS)の表面電位測定により詳しく測定されている35。さらに、CuPcをホール注入層として15 nmほど挿入することによって、ホール注入特性が改善される。CuPcについては、酸素吸着による導電率の変化や、膜厚によるモルフォルジーの変化も知られていることから、UPS測定による詳細な陽極解析を行う必要がある36。逆に陰極では、仕事関数の小さな金属電極から電子輸送層の最低空軌道(Lowest Unoccupied Molecular Orbital:LUMO)に電子が注入される。Alq3のLUMOは、-3.3 eV程度であり、-3.8 eVの仕事関数を持つMg0.9Ag0.1合金と、Alの仕事関数-4.3 eVと比べるとMg0.9Ag0.1の方が陰極に有利であるが、デバイスの作製プロセスの関係からAlの方が好まれる。その場合、バッファ層として、アルカリ金属やアルカリ土類金属の酸化物やハロゲン化物が極薄膜で用いられる。代表的なバッファ層が、LiFであり、電子輸送層とAl電極間に0.5 nmから1.0 nmのLiF層を挿入すると電子の注入効率が改良され、効率もMg0.9Ag0.1よりも向上することが知られている22。さらに、これまでに成功を収めたCuPcやLiF の極薄膜の有機ELへの挿入は、電極と有機物の電子状態のマッチングだけでなく、表面状態の改質にも大きな影響を与えると考えられ、有機EL素子の駆動寿命を大きく向上させている38。

図2-2 2層型有機EL素子のエネルギー準位図
キャリアの移動過程
注入されたキャリアは有機層内を移動するが、有機EL素子に用いる材料のキャリア移動度は10-3~10-6 cm/V・s程度であり、アモルファス膜を利用していることを考慮すると、分子間でのπ-π相互作用はそれほど強固ではなく、明確なバンド構造を形成しているとは考えにくい。そのため、ホールと電子の移動は、中性分子とそれぞれアニオンラジカルやカチオンラジカルをやりとりすることで、分子間をホッピングによって移動していると考えられている。ここで、有機EL素子の電導機構として提案されている空間電荷制限電流(Space Charge Limited Current, SCLC)39を検討してみる。元来、キャリアを薄膜中に持たず、キャリア移動度が小さな有機薄膜の電気伝導はSCLCと考えられている。電流密度J(A/cm2)は、キャリア移動度μ(cm/V/s)と膜厚L(m)、電圧V(V)と誘電率ε(F/m)の関係式として次式で示される。
 (2.1)
(2.1)
そこで、膜厚を100 nm、有機薄膜の移動度を10-3 cm/V・s、十分に有機EL素子から発光が観察される電圧10Vとし定数項を代入すると、有機薄膜中を流れる電流密度はおよそ30 A/cm2となり、移動度の小さな有機薄膜でも膜厚を十分に薄くすることによって大電流を流せることが分かる。
キャリア再結合と輻射失活過程
発光中心上でホールと電子は、クーロン力によって再結合し、励起子を生成する。その際、スピン多重度の違いにより発光性の一重項励起子と非発光性の三重項励起子が1対3の割合に分配される(図2-3)ことが知られている27。つまり、電気的に励起された励起子のうち、75%生成される三重項励起子からのリン光は禁制であり、非輻射失活により発光として取り出せず熱になってしまう。ただし、前述したようにIr錯体を用いた有機EL素子のようにリン光発光デバイスを用いた場合は、一重項から三重項への項間交差(Intersystem Crossing:ISC)も十分効率よく起こると仮定すると、生成した励起子を100%発光に利用できる可能性がある。

図2-3 一重項励起子と三重項励起子
光取り出し過程
最後に、有機層中で発生した光は、屈折率の低い空気中へ取り出されて我々の目に認識される。古典光学から単純に導いた有機EL素子からの光取り出しの効率は、20%程度と見積もられ、残り80%の光は、基板を導波したり金属電極に吸収されたりするなどして消失してしまうと考えられている40。最近では三次元の時間領域有限差分法(Finite-difference time-domain:FDTD)41による発光のモード解析によっても同様の値が算出されている。改善方法として、素子一つ一つをメサ型構造の基板上に作製することや42、マイクロレンズをつける43ことによってガラス基板を導波する光を取り出す試みが行われている。また、ガラス基板も導波しないようにITOを加工しITOと有機層を導波する光を取り出す44手法も考案されている。有機EL素子の開発において、光取りだし効率の決定的な解決策は見つかっておらず最も大きな課題の一つである。
以上のような素過程の各効率から最終的に有機EL素子の量子効率が算出される。つまり、有機EL素子に注入されたキャリア数に対する、素子から取り出したフォトン数で定義される外部量子効率ηφ(ext)は次式で表され、有機EL素子の特性を評価する重要な指針となっている。
 (2.2)
(2.2)
それぞれの変数に理論的な効率を代入すると、外部量子効率の最大値は、蛍光を利用した素子において5%、リン光を活用した素子で20%となる。(詳しくは「AppendixⅠ §A1-2. 量子効率の詳細な検討(PDF版)」を参照)
§2-2.有機EL素子構造
§2-2-1.基本的な有機ELの素子構造
有機ELの基本的な素子構造として、下記3つを挙げた。層構造を表すのに、物質名(膜厚)を陽極から陰極へ順番に記載するのが一般である。
- 最も基本的な構造である、C. W. Tang 等の提案した二層積層型素子(図2-2参照)
ITO / α-NPD(50 nm)/ Alq3(50 nm)/ MgAg(150 nm)/ Ag(10 nm)
(陽極 / ホール輸送層 / 電子注入輸送層兼発光層 / 陰極) - 発光効率を向上させ、発光色を制御するために蛍光量子効率の高いDCMやクマリンをドーピングした素子
ITO/ α-NPD(50 nm)/ Alq3:1mol% - DCM(50 nm)/ MgAg(150 nm)/ Ag(10 nm)
(陽極 / ホール輸送層 / ホスト材料-ドーパント(発光層)/ 陰極) - リン光発光材料を用いた素子
ITO / TPD(40 nm)/ CBP:6wt% - Ir(ppy)3(20 nm)/ BCP(10 nm)/ Alq3(30 nm)/ LiF(0.5 nm)/ Al(100 nm)
(陽極 / ホール輸送層 / 発光層 / ホールブロック層 / 電子注入層 / 陰極)
素子構造は、材料のキャリア輸送性やHOMO、LUMO準位から予想して設計することもできる。2.で用いられているDCMは、Alq3よりもバンドギャップが小さくAlq3から効率よくエネルギー移動し、DCMが発光する良い材料である(図2-4)。しかし、DCMの単層膜ではDCMが濃度消光してしまうために、1 mol%程度の低濃度でホスト材料にドーピングして用いる。なお、一般的に陰極には、MgAg合金もしくはLiF / Alの積層電極を用いることが多い。

図2-4 ドーピング型における発光層のエネルギー準位図
リン光発光有機ELの素子設計は、HOMO-LUMO準位から検討しただけではうまくいかないことが多い。それは、蛍光スペクトルや紫外可視吸収スペクトル、UPSなどより決定したHOMO-LUMO準位は、一重項励起状態に対応するエネルギー準位の指標であり、リン光発光有機ELでは、三重項励起状態に対応するエネルギー準位で検討しないとならないためである。520 nm程度にリン光を発するIr(ppy)3にとって、発光スペクトルに重なる波長域にα-NPDの三重項に由来する吸収帯が存在するため、その領域に吸収帯を持たないTPDを用いた方が効率は高くなる。
§2-2-2.有機ELを構成する基本的な材料
ホール注入材料(製品リスト)
ホール輸送層のHOMO準位と陽極の仕事関数との間にHOMO準位を有し、陽極から有機層への掘る注入障壁を下げる働きをする。

ホール輸送材料(製品リスト)
発光層へホールを輸送する働きをし、発光層と接するため発光層から励起エネルギーが移動せず、さらには高層と相互作用してエキサイプレックスを形成しないように、発光層よりもエネルギーバンドギャップが大きな材料が用いられる。

電子輸送材料(製品リスト)
陰極から電子を注入し輸送する機能を持つ。ホール輸送層と同様に、バンドギャップが広い材料が好ましい。また、発光層内で生成した励起子の移動を阻止する働きもあるため、励起子阻止層や、BCPはホールの移動を阻止する作用があるため、ホールブロッキング層として使われることもある。

発光材料(製品リスト)
発光材料として最も有名なものは、Alq3であり、ホール輸送層と組み合わせて用いられる。その他にも、金属錯体には電子輸送性を併せ持つ発光材料も多く発表されている。発光材料の中でも、高濃度条件下で蛍光量子収率が減少(濃度消光)する材料は、ホスト材料に発光材料を分散させて用いる。このような材料には、希薄状態で100%近い蛍光量子収率を示すレーザー色素材料であるCoumarinやDCM、ルブレン等がある。
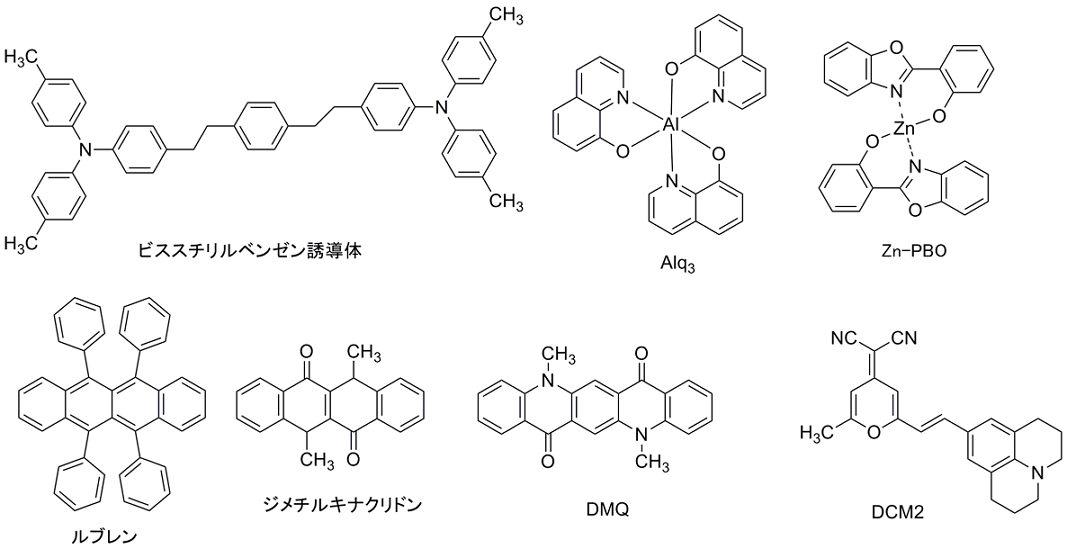
ホスト材料(製品リスト)
発光材料の濃度消光が激しいときや、発光材料のキャリア移動度が遅く単層膜として挿入できない場合など、バイポーラー性のホスト材料中に発光色素(ゲスト材料)をドーピングすることがある。ホスト材料は、ゲスト材料よりも大きなバンドギャップを有している必要がある。また、リン光発光材料をドーピングするときは、ホスト材料の三重項のバンドギャップもリン光発光材料よりも大きい必要があり、小さい場合はエネルギー移動し、エネルギーを閉じ込められなくなったり、ホスト材料の三重項から熱失活してしまったりするので、材料の選択には注意を要する。

燐光(リン光)材料(製品リスト)
リン光材料には、禁制である三重項からの発光を得るため、重原子効果を利用する。そのため、中心金属に白金やイリジウムを有する材料が報告されている。これらの金属錯体は、配位子のπ電子の広がりを制御することによって、青色~赤色の発光色が得られている。

高分子材料(製品リスト:ホール輸送材料、発光ポリマー)
高分子材料は、欧米を中心に開発されてきた歴史がある。最も基本的なポリフェニレンビニレン(PPV)を基本骨格として、アルキル基を付加することにより、溶解性を向上させたり、置換基を付加することにより、発光色やキャリア輸送性を制御して開発が進んだが、PPVは黄色発光をするため、青色発光材料の開発が困難であった。しかし、固体薄膜状態で高効率に青色発光を示し、熱的にも化学的にも安定であるフルオレン系高分子が見いだされ、高分子有機ELにおいても、青色~赤色に渡る発光が得られている。

§3.有機ELの作製方法
§3-1.材料の昇華精製
有機半導体デバイスに用いられる材料には、長寿命化や高信頼性のため高純度化が要求される。最近では、有機半導体用に昇華精製された材料も入手できるようになっている。特に、真空中であっても加熱により有機材料の分解を促進する触媒作用を持つ不純物や、チャンバー内や真空度を汚染する残留溶媒などの除去は非常に重要である。一般に、有機材料はカラムクロマトグラフィーや再結晶を繰り返して精製される。しかしながら、残留溶媒の完全な除去は困難であり、難溶性の材料には適用しにくい欠点を有する。そこで、真空蒸着に用いる有機材料は、必ずといって良いほど昇華精製を行い、十分に不揮発性の不純物や目的材料よりも低沸点な残留溶媒や不純物を取り除くようになった。加熱すると蒸発または昇華する(以後、昇華と表記)物質は、真空雰囲気にすることにより、昇華温度は低下するが、C-C結合やC-H結合を熱エネルギーによって切断することに相当する材料の分解温度は低下せず、さらに真空下では酸素濃度も低くなるために酸化も起こりにくくなり、分解することなく材料を昇華させることができる。
昇華精製法は、図3-1に示すように、石英管、石英管を取り巻く温度勾配形成用金属管、その金属管に設置されたバンドヒーター、温度調節器、(ターボ分子)ポンプで構成される。粗精製材料をポンプとは反対側のヒーター部に数g置き、石英管内を10-3 Pa程度の真空状態に保つ。ヒーターで材料を仕込んだ一端を加熱することになるので、管内に温度勾配ができる。このため、温度勾配形成型昇華精製装置は、Train Sublimation型精製装置とも呼ばれる。有機材料が蒸発するまで加温すると、蒸発した有機材料は熱拡散とポンプに引かれて低温側へ移動する。この時、ヒーター側からキャリアガスを導入することもある。そして、温度勾配により、純物質と不純物の析出温度差を利用し、不純物を分離する方法である。
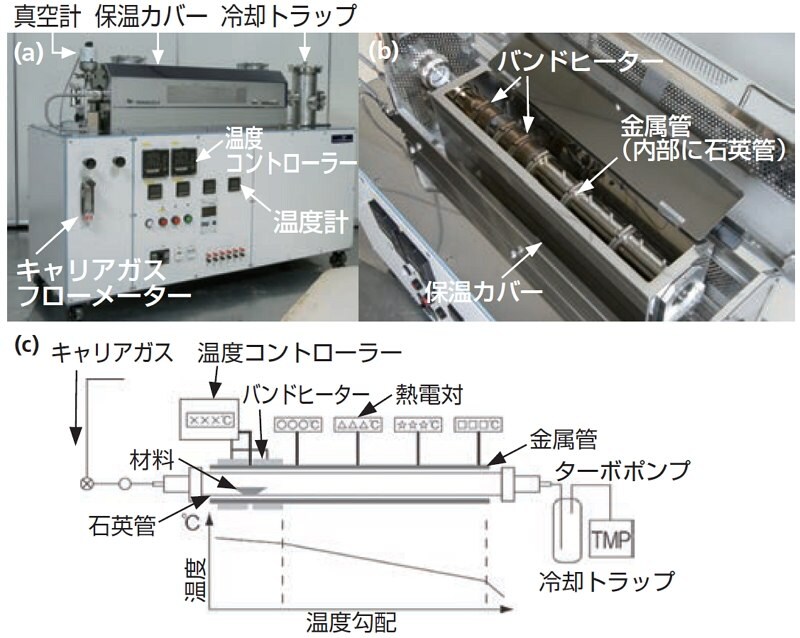
図3-1 Train Sublimation型精製装置の写真と概略図(a)全体写真(b)装置内部写真(c)概略図
不揮発性の不純物は原料ボートに残り、分子量の大きな分子は高温側に、低分子量の不純物は低温側に分離され、しばしば不純物と精製物が帯状に析出する(図3-2)。カラムクロマトグラフィーが、充填剤と純物質と不純物との吸着力差を利用しているのに対して、昇華精製は析出温度差を利用しているため、カラムクロマトグラフィーの乾式工程と考えることもできる。このため、残留溶媒なども効果的に取り除くことができ、物質の昇華性を判断することも可能であり、最終的な精製法として優れている。通常は、不純物の析出が見られなくなるまで繰り返し昇華精製を行う。しかし、析出温度が非常に近い物質を分離することは原理的に困難である。また、この温度勾配形成型昇華精製装置はキャリアガス種や真空度を調節すれば、装置構成を変えることなく単結晶育成装置としても転用することができる。

図3-2 CuPcの昇華精製
なお、図3-3の様な方式でも昇華(精製)に変わりないが、解説したTrain Sublimationの精製原理より、単純に昇華させた材料を回収するだけの方法では有機デバイス作製に必要十分な精製はできないと考えられ、材料購入の際などにはどの方式で昇華精製された材料なのか注意が必要である。

図3-3 単純な昇華精製装置の概念図
§3-2.ITO基板の取り扱い
有機ELの実験に用いるITO(Indium-Tin Oxide)透明電極基板は、面抵抗10 Ω/□のガラス基材品が比較的入手しやすく、初期特性評価などには全く問題ない。そのITO基板はエッチングを行う方法、及び、そのまま使う方法とどちらでもかまわないが、ここでは、まずエッチングの方法について述べる。さらに、複雑なパターンが必要なときには、フォトレジストを用いてパターンを形成するが、フォトレジストの使用法については解説が多くあるので他書に譲り、最も容易にできるメンディングテープを使ったエッチング法について述べる。
§3-2-1.ITOのエッチング
- メンディングテープを直接基板に貼り付け、不要な部分を取り除き、マスキングをする。

- ドラフト中で、王水(塩酸:硝酸= 1:3 vol ratio)をガラスシャーレに調整し、ITO面を上にしてマスキングしたITO基板を浸す。このとき、エッチング液は、市販のものを使用しても良い。
- およそ10分でエッチングが完了するので、王水に注意しながらプラスチックピンセットを用いてITO基板を取り出し、ビーカーにとったイオン交換水で十分にすすぎ、さらに流水ですすぐ。
- ITO基板についた水分をよく拭き取り、テスターでITOがエッチングされているか確認し、テープを粘着質が残らないようにゆっくりと剥がす。
- この操作以降は、基板の洗浄を行うが、ウオーターマークとして洗浄しても落ちにくい汚れがつくのを防ぐため、ITO基板が乾燥しないように注意する。
§3-2-2.基板洗浄
- 図3-4のようなテフロン製基板ホルダーに基板をたててビーカーに入れ、理化学機器用の中性洗剤で5分、イオン交換水で洗剤が落ちるまで5分を数回、アセトン、イソプロピルアルコール(IPA)を用いてそれぞれ二回ずつ5分間超音波バス中で洗浄を行う。洗剤は、ITO専用のものも販売されているので、それを利用しても良い。
- 洗浄がすんだITO基板は、乾燥しないように新しいIPAもしくはエタノール中に保存する。
- 有機ELの作製直前に洗浄したITO基板を、IPAかエタノールを半分ほど満たしたビーカーに入れ、突沸しないように加熱し、沸騰させる。
- 綺麗に洗浄したピンセットを用いて、ゆっくりと引き上げて乾燥させる。
- さらにUV/オゾン洗浄を10~15分間行う。
- すぐに真空蒸着装置にセットし、有機半導体材料を成膜する。

図3-4 基板洗浄ホルダー
§3-3.真空蒸着装置を用いた有機ELの作製
§3-3-1.真空蒸着装置
今では、真空蒸着装置があれば、有機ELの作製は簡単に行うことができる。ここではまず、真空を利用して薄膜作製を行う上で重要な基本概念である平均自由行程と入射頻度について述べる。平均自由行程 λ(m)とは、ある圧力下で残存する気体分子と衝突することなく分子が飛ぶことのできる距離の平均値を表し、圧力P(Pa)、温度T(K)および分子の直径D(m)との間には次のような関係がある45。
λ = 3.1×10−24 T / PD2 (3.1)
ここで、有機ELに発光層として一般的に用いられるAlq3を例に計算してみる。参考文献46からAlq3の分子密度は2×1021個/cm-3程度なのでAlq3分子を球と仮定すると、Alq3の直径をおよそ1.4 nmと算出できる。室温(300K)で製膜した時、1 Paでの λ は~0.5 mm、10-3 Paではλ は~50 cmとなる。しかし、平均自由行程はあくまで「平均値」であることを考慮すると、10-3 Paで蒸着源から熱エネルギーを受け、飛び出した高いエネルギー状態にある分子が、途中酸素などの残留気体分子と衝突せずに50 cm直進できる割合は、わずか3割に満たないため、蒸着源と基板間距離が30 cm程度ある一般的な真空蒸着装置では、10-4 Pa以上の高い真空度に保つことが必要になる。また、入射頻度Zn(個/m2・s)は、基板に衝突する気体分子の頻度を表す。圧力P(Pa)、温度T(K)、気体の分子量M(g/mol)の間には次の関係がある45。
Zn = 2.6 ×1024 P/(MT)1/2 (3.2)
10-4 Paにおいて300 Kでの残存酸素(M02=32)の入射頻度は、2.7×1018個/m2程度となる。先に算出した直径1.4 nmのAlq3分子が基板最表面に単分子層を形成したとすると、基板表面には約6.5×1017個/m2の分子が並んでいることになるので、1秒も経たないうちに表面にあるすべての分子が酸素分子の衝突を受けてしまうことになる。そのため、より高真空状態の実現が必要となることがわかる。
また、有機半導体分子は酸素存在下で加熱すると、酸化反応が進み炭化してしまうことが多い。しかし、高真空下では沸点降下現象により沸点(昇華点)は低下するが、有機分子を構成するC-C結合などの化学結合を解離・分解するエネルギーは影響を受けない。そのため、大気中で分解することなく昇華(蒸発)することができない有機半導体材料も、酸素も取り除かれた高真空状態で加熱することによって、容易に昇華させ基板上へ薄膜を製膜することが可能となる。
以上のように、有機半導体デバイスの作製には真空度は高ければ高いほど好ましいが、一般的には、比較的簡便な装置構成で、清浄な雰囲気を実現できる10-4 Pa程度の真空度で製膜を行うことが多い。しかし、製膜時の真空度は、10-3 Pa以上の高真空下ではデバイスの初期特性にはほとんど影響を与えないものの、製膜時の雰囲気によって有機材料の結晶化が促進されるH. Azizらの報告や47、製膜時にデバイスに取り込まれた酸素や水分が、駆動に伴う電圧の印加によってデバイスを構成する材料と不可逆な電気化学的な反応を引き起こすため、駆動寿命へ大きな影響を与えるとするT. Ikedaらの報告48などに留意する必要がある。
真空蒸着には10-4 Paオーダーの真空度を得るため、安価で比較的メンテナンスも容易な油拡散ポンプとロータリーポンプを組み合わせた排気系が数年前まで用いられていた。しかし、オイルミストや熱分解したオイル成分が、きわめてわずかであるが真空チャンバー内へ混入して不純物として振る舞うために、現在では、水分を効果的に除去できるクライオポンプや、メンテナンスがほぼ必要のないターボ分子ポンプと液体窒素トラップを組み合わせたドライな排気系が主流となっている。また、真空ポンプだけではなく、真空チャンバーの構成部品には、シャッターの開閉や基板回転機構、基板搬送機構など、直線運動や回転運動を必要とするものがある。これらの機構にも、ベローズや磁気結合式駆動伝達機構を設け、潤滑油を必要としない装置構成とするなどの注意が必要である。
蒸着装置は、1台でも有機デバイスを作製することは可能であるが、有機物用と金属用に2台の装置を準備し、ゲートバルブで接続することが好ましい。これは、融点の高い金属電極を製膜する際、金属材料の加熱による輻射熱の影響を受け、チャンバー内部に付着した有機物が再蒸発し、コンタミネーションの原因となることを防ぐためである。図3-5にマルチチャンバー型有機デバイス製膜装置の概念図を示した。最近では、チャンバー間をゲートバルブで接続し、真空搬送機構を設け、基板投入から製膜、グローブボックスまで一度も大気に曝すことなくデバイスの作製と封止を行える装置構成が一般的になりつつある。真空一貫で作製したデバイスを、大気に曝すことなく酸素や水分濃度が0.1 ppm以下のグローブボックスへ取り出し、ガラスやアルミ缶などのガスバリア性の高い封止缶とUV硬化樹脂を用いて封止まで行うことは、デバイス作製直後の初期特性に影響を与えることは少ないが、デバイスの長寿命化や信頼性確保のために必要となる。特に、雰囲気に非常に敏感な有機FETデバイスに関する研究には必須の装置構成となる。有機FETは、半導体層への酸素や水の吸着、半導体/絶縁層界面の状態によって半導体層中のキャリアトラップ密度が変化し、このキャリアトラップが素子性能(FET移動度)に大きな影響を与えると考えられている。

図3-5 マルチチャンバー型有機デバイス製膜装置の概略図
次に、真空成膜装置内部(図3-6)の主要な構成について述べる。現在の有機ELは、高度に機能分離された積層構造やドーピングが必要不可欠なため、多元蒸着源が必須となる。さらに、限られた空間の中に蒸着源を多数設置することから、互いに干渉、及び、相互の蒸着源を汚染させないためにも蒸着源の間は仕切板で分離される必要がある。また、2種類の有機材料を共蒸着法により製膜できるように、2つの蒸着電源及び膜厚計がホスト材料用とゲスト材料用にそれぞれ必要となる。実際には、基板近傍に設置されたホスト用膜厚計に、ゲスト材料が入射しないようにすることは困難であり、基板用膜厚計と同一になるように設計しても良い。さらに、蒸着源は輻射熱の影響を避け、基板近傍で有機材料の蒸気を均一化するためにも、経験的に基板から30 cm以上離れた位置に配置する。しかし、蒸着源が複数設置される場合には、蒸着源を基板直下に集中することができないため、膜厚のムラが危惧されるが、10~12 rpm程度の速度で基板を回転させるだけで、10 cm角の基板を用いても、膜厚やドーピング濃度のムラを数%以下まで抑えることができる。
有機材料の蒸着源には、Ta、Mo等金属製の昇華ボートに材料を投入し、金属製ボートに電流を流し、その金属の抵抗により発熱させる単純な抵抗加熱方式や、石英や黒鉛、BN等でできたるつぼをタングステンヒーターで加熱する簡易K-セルタイプが一般的に用いられる。しかし、一般的に有機材料の熱伝導率は悪く、加熱されやすい蒸着ボート壁面から材料は蒸発するため、有機材料の突沸やボート内での材料の崩落により蒸着速度が大きく変化することがある。最近では、有機材料とともにサーモボールと呼ばれる化学的に安定な良熱伝導性の無機材料を混合して加熱することにより、蒸着速度を安定化させる手法も提案されている。

図3-6 典型的な有機蒸着室の内部
金属の蒸着源にも様々なボートが市販されているが、蒸着ボートと合金を作る金属もあり、ボートの材質の選択が必要なものがある。有機デバイスに一般的に用いられるMgやAg、Ca、電子注入層に用いられるLiFなどは、WやTaと合金を作らないため、粉末状の材料であればボックス型ボート、ある程度の固まり状であれば、安価なV字型W製ボートなどが便利である。ところが、Alは様々な金属や酸化物と合金を作るため、蒸着には困難を伴う。そのため、Alの蒸着は、電子ビーム蒸着法を用い、ハースライナーを使用せず十分に水冷されたCu製るつぼに直接投入し、E-型電子銃で加熱し蒸発させるか、Alと合金を形成するもののW製スパイラルボートを使い捨てで用いる。Li、Csなど活性の高いアルカリ金属は、アルカリディスペンサーを用いることが多い。蒸着電源は、共蒸着ができるように、有機、金属用共に2式以上準備することが望ましい。抵抗加熱方式の場合、蒸着電源は10 V、100 A程度の出力パワーがあれば、PtやTaなど高融点金属材料を除いたほとんどの金属材料や450℃以上の昇華温度を有する有機物も蒸着できる。図3-7に様々なタイプの蒸着ボートを掲載した。

図3-7 様々な蒸着ボートの種類と一部の断面図
真空蒸着の場合は、蒸発源の電流(もしくは蒸着ボートの温度)を一定に保持しても蒸着速度は一定になることはない。また微量のゲスト材料をホスト材料に共蒸着させるには、精密に蒸着速度を制御しなければならない。そのため、膜厚の測定には、真空下で使用でき、オングストロームオーダーの膜厚計測が可能な水晶振動子式膜厚計(図3-8)を用いる。
水晶振動子は広く時計に使われており、その固有振動数は非常に安定している。このような性質を持つ水晶振動子に交流電場を印加すると、水晶振動子の固有振動数と交流電場の振動数が等しくなったところで共振現象が起こる。この水晶振動子表面に物質が蒸着されると、水晶振動子の固有振動数は低い振動数の方向に変化する。この変化量は蒸着物質の質量に比例する。つまり、共振周波数の変化を精度よく検出すれば、蒸着物の付着質量を膜厚に換算して膜厚が測定できることになる。水晶振動式膜厚モニターは、蒸着物の密度を入力し、Z-ratioと呼ばれる水晶振動子と蒸着物質の音響インピーダンスの補正を行うパラメーターを入力、さらに、触針式の膜厚計やエリプソメーターによって膜厚を実測し、水晶振動子式膜厚計のモニター値のズレを補正(Tooling Factor)する必要がある。実際に水晶振動子に付着した蒸着物の質量を、オングストロームオーダーの膜厚として検出できる水晶振動子式膜厚計は、水晶振動子に入射する蒸着材料の量に非常に敏感になるため、膜厚補正した水晶振動子検出器の位置や角度の固定には十分に注意を払い、定期的にTooling Factorの再補正を行う必要がある。
一般的な有機EL素子を作成する場合、素子を構成する各膜の標準的な厚さ(数10~100 nm前後)であれば、装置の大きさにも依存するが、有機半導体材料はそれぞれおよそ30 mg程度、さらにゲスト材料では10 mg程度蒸着ボートに仕込めば十分である。

図3-8 水晶振動子式膜厚計の配置
§3-3-2.ドーピングの手法
ドーピング(共蒸着)法の蒸着速度の算出法と実験レベルでの複数の膜厚計の使用法について述べる。ここで、ゲスト材料をX mol%の濃度でホスト材料にドープしたい場合、ホスト材料の分子量をMhost、質量をWhost、蒸着速度をRhost、ゲスト材料の分子量をMgest、質量をWgest、蒸着速度をRgestとしたとき、ホスト材料の蒸着速度に対するゲスト材料の蒸着速度は、以下のように計算できる。
 (3.3)
(3.3)
 (3.4)
(3.4)
水晶振動子では、単位時間あたりの質量を測定しているので、
 (3.5)
(3.5)
 (3.6)
(3.6)
上式より、ホストとゲスト材料の分子量が既知であり、設定したい濃度(X)を決めると「比」の形で表すことができる。有機ELでは、ホスト材料の蒸着速度がおよそ1~5 Å/s内に収まるよう設定することが多いので、適当な値を代入することによりゲスト材料の蒸着速度を求めることができる。言うまでもないが、重量比(wt%)でドープ濃度を設定する場合には、蒸着速度比をそのまま重量比として読み取ればよい。ここで、分子量Mhost=459.44 g/molのAlq3へ、Mgest=303.36 g/molのDCMを1 mol%ドープする場合の膜厚計の使い方について考えてみる。式(3.6)より、
Rgest = 6.67 × 10-3 × Rhost (3.7)
となるので、仮にAlq3を基板用膜厚計でRhost=5 Å/sの蒸着速度となるように成膜することを考えると、同じく基板用膜厚計でRgest=0.033 Å/sの蒸着速度になるように制御する必要がある。つまり、10秒間に0.33 Åの蒸着速度に相当するが、膜厚計の有効数字から考えてもまだ遅すぎるため調整ができない。この時Tooling FactorがY%であるならば、モニターのパラメーター入力値を3 × Y %と3倍してみる。すると、見かけ上の蒸着速度は、Rhost=15 Å/s、同じくRgest=0.099 Å/s、つまり、10秒間に~1 Å成膜されるように制御すればよいことになる。そこで、Tooling Factorを3倍にした基板側膜厚計でRgest~1 Å/10sとなるように、ゲストの蒸着速度を調節し、この時のゲスト側膜厚計での蒸着速度を読み取る。この時、基板側の膜厚計とゲスト側の膜厚計には比例関係が成立しているので、ゲスト側膜厚計で膜厚補正をする必要はなく、パラメーターも蒸着速度を読み取りやすいように任意の値でよい。この後、ホスト材料の蒸着を始めると、基板側でゲスト材料の蒸着速度が制御できなくなることから、ゲスト材料の蒸着速度の管理は、ゲスト側膜厚計での読み値を参考に制御する。このゲスト用の読み値は、蒸着ボートのわずかな設置位置のズレによって、大きく変化するので実験ごとに取り直した方がよい。ゲスト材料の蒸着速度を厳密に制御しながら、ホスト材料の蒸着速度調整を始め、見かけ上Rhost=15 Å/sになるように調整して成膜する。この時、Tooling Factorを3倍しているので、実際に基板上へ成膜された膜厚はモニター出力値の1/3になっていることに注意する。同様の考え方で、Mg:Ag(10:1 wt ratio)も成膜できる。
§3-4.特性評価
有機EL素子の特性評価は、電圧を印加しそのときの電流及び発光強度とEL(発光)スペクトルを測定する必要がある。プログラマブルな電源と光パワーメーター、及びマルチチャンネルアナライザーがあると、比較定容易に測定プログラムも作れ、測定もできる。電源は、20 Vも出力できればよく、電流計は電源内蔵の装置が入手できる。このとき、有機EL は、電流の測定レンジが広く必要なため、1 pA~100 mA、できれば1 A程度まで測定できる装置が好ましい。また、光量測定においては、高価な輝度計でもよいが、nWからmWオーダーで測光できるフォトダイオードをディテクターに用いた可視域用光パワーメーターがあれば十分であり、輝度(cd/m2)、外部量子効率(%)、電力効率(lm/W)等は容易に算出できる。これらの機器を接続し、電圧ステップを0.1V~0.5V幅で、15V(おそらく破壊される)程度まで印加し、そのときの電流値及び発光強度を同時に測定する。
§4.データー整理
(PDF版をご覧ください)
§5.おわりに
以上「有機EL素子の基礎及びその作製技術」では、学術論文や解説書等では詳細に記述されない「実験」について、可能な限り一般化して紹介した。また、データの整理法や、外部量子効率の算出法なども詳しく解説した。本稿が、有機ELの研究をする方の一助になれば幸いである。また、有機ELの動作原理や、素子設計については「有機ELディスプレイ(時任静士、安達千波矢、村田英幸 共著、オーム社)」等の良書も多く出版されているので参考にして欲しい。Appendix(PDF版)には、本編で取り上げられなかった語句の説明や、スペクトルを考慮した外部量子効率の算出法も記載したので参考にして欲しい。末筆ながら、本稿の執筆を勧めていただいたシグマ アルドリッチ ジャパン(株)吉田氏に感謝する。
昇華グレード製品
References
- A. Bernanose, M. Conte, P Vouauzx, J. Chim. Phys., 1953, 50, 64.
- M. Pope, H. P. Kallmann, P. Magnante, J. Chem. Phys., 1963, 38, 2042.
- W. Herfrich and W. G. Schneider, Phys. Rev. Lett., 1965, 14, 229.
- W. Herfrich and W. G. Schneider, J. Chem. Phys., 1965, 44, 2902.
- W. D. Gill, J. Appl. Phys., 1972, 43, 5033.
- G. Pfi ster, Phys. Rev., 1977, B 16, 3676.
- P. M. Borsenberger, W. Mey and A. Chowdy, J. Appl. Phys., 1978, 49, 273.
- P. S. Vincett, W. A. Barlow and R. A. Hann, Thin Solid Films., 1982, 94, 171.
- G. G. Roberts, M. M. McGinniity, W. A. Barlow, P. S. Vincett, Solid State Commun., 1979, 32, 683.
- S. Hayashi, T. T. Wang, S. Matsuoka, S. Saito, Mol. Cryst. Liq. Cryst., 1986, 135, 355.
- C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913.
- C. Adachi, S. Tokito, T. Tsutsui and S. Saito, Jpn. J. Appl. Phys., 1988, 27, L269.
- C. Adachi, S. Tokito, T. Tsutsui and S. Saito, Jpn. J. Appl. Phys., 1988, 27, L713.
- C. Hosokawa, H. Higashi, H. Nakamura, T. Kusumoto, Appl, Phys. Lett., 1995, 67, 3853.
- Y. Hamada, T. Sano, M. Fijita, T. Fujii, Y. Nishio and K. Shibata, Jpn. J. Appl. Phys., 1993, 32, L514.
- Y. Shirota, Y. Kuwabara and H. Inaba, Appl. Phys. Lett., 1994, 65, 807.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539.
- Y. Ohmori, M. Uchida, K. Muro and K. Yoshino, Jpn. J. Appl. Phys., 1991, 30, L1938.
- Y. Ohmori, M. Uchida, K. Muro and K. Yoshino, Jpn. J. Appl. Phys., 1991, 30, L1941.
- A. R. Brown, D. D. C. Bradley, J. H. Burroughes, R. H. Friend, N. C. Greenham, P. L. Burn, A. B. Holmes and A. Kraft, Appl. Phys. Lett., 1992, 61, 2793.
- J. Kido, H. Shionoya and K. Nagai, Appl. Phys. Lett., 1992, 67, 2281.
- T. Wakimoto, Y. Fukuda, K. Nagayama, A. Yokoi, H. Nakata, M. Tsuchida, IEEE Transactions Electron Devices, 1997, 44, 1245.
- N. Takada, T. Tsutsui and S. Saito, Appl. Phys. Lett., 1993, 63, 2032.
- 山本幸之助,筒井哲夫,表面,1998, 36, 479.
- 安達千波矢,日本学術振興会EL分科会第19回研究会資料,p18.
- M. A. Baldo, S. Lamansky, P. E. Burrows, M. E. Tompson, S. R. Forrest, Appl. Phys. Lett., 1999, 75, 4.
- M. A. Baldo, D. F. O’ Brien, Y. You, A. Shoustikov, A. Aibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151.
- T. Tsutsui, M. Yang, M. Yahiro, K. Nakamura, T. Watanabe, T. Tsuji, Y. Fukuda, T. Wakimoto, S. Miyaguchi, Jpn. J. Appl. Phys., 1999, 38, L1502.
- 森川通孝,安達千波矢,筒井哲夫,斎藤省吾,第51回応用物理学会学術講演会予稿集,1990, p1041.
- S. Hoshino and H. Suzuki, Appl. Phys. Lett., 1996, 69, 224.
- C. Adachi, M. A. Baldo, S. R. Forrest and M. E. Tompson, Appl. Phys. Lett., 2000, 77, 904.
- C. Adachi, M. A. Baldo, S. R. Forrest, S. Lamansky, M. E. Thopmson and R. C. Kwong, Appl. Phys. Lett., 2001, 78, 1622.
- C. Adachi, R. C. Kwong, P. Djurovich, V. Adamovich, M. A. Baldo, M. E. Thompson and S. R. Forrest, Appl. Phys. Lett., 2001, 79, 2082.
- S. Tokito, T. Iijima, Y. Suzuki and F. Sato, Appl. Phy. Lett., 2003, 83, 569.
- K. Sugiyama, H. Ishii, Y. Ouchi and K. Seki, J. Appl. Phys., 2000, 87, 295.
- 佐藤佳晴,有機EL材料とディスプレイ,第7章,2001, p.103.
- T. Wakimoto, Y. Fukuda, K. Nagayama, A. Yokoi, H. Nakada and M. Tsuchida, IEEE Trans. Electron Devices, 1997, 44, 1245.
- Y. Sato, T. Ogata, S.Ichisawa, M.Fugono and H. Kanai, Proc. SPIE, 1999, 3797, 198.
- P. E. Burrows, Z. Shen, V. Bulovic, D. M. McCarty, S. R. Forrest, J. A. Cronin and S. R. Forrest, J. Appl. Phys., 1996, 79, 7991.
- N. C. Greenham, R. H. Friend and D. D. C. Bradlay, Adv. Mater., 1994, 6, 491.
- チュティナン・アロンカーン,冨士田誠之,石原邦亮,国師渡,上野哲也,浅野卓,野田進,第50回応用物理学関係連合講演会予稿集,No3, 2003, 1408.
- G. Gu, D. Z. Garbuzov, P. E. Burrows, S. Venkatesh, S. R. Forrest and M. E. Thompson, Opt. Lett., 1997, 22, 396.
- S. Moller and S. R. Forrest, J. Appl. Phys., 2002, 91, 3324.
- M. Fujita, T. Ueno, T. Asano, S. Noda, H. Ohhata, T. Tsuji, H. Nakada, N. Shimoji, Electron. Lett., 2003, 39, 1750.
- 麻蒔立男,薄膜作成の基礎,日刊工業新聞社,東京,1977.
- P. E. Burrows, Z. Shen, V. Bulobic D. M. McCarry, S. R. Forrest, J. A. Cronin, M.E. Tompson, J. Appl. Phys., 1996, 79, 7991.
- H. Aziz, Z. D. Popovic, S. Xie, A. M. Hor, N. X. Hu, C. Tripp, G. Xu, Appl. Phys. Lett., 1998, 72, 756.
- T. Ikeda, H. Murata, Y. Kinoshita, J. Shike, Y. Ikeda, M. Kitano, Chem. Phys. Lett., 2006, 426, 111.
続きを確認するには、ログインするか、新規登録が必要です。
アカウントをお持ちではありませんか?