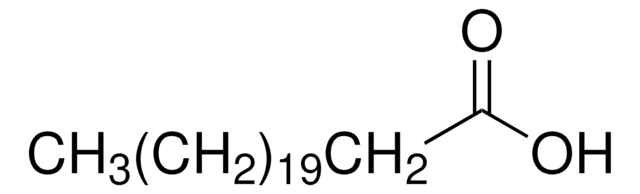Oxidation product of fatty acids in the diet, particularly phytanic acid. Phytanic acid is oxidized by alpha oxidation yielding pristanic acid, which is subsequently degraded by peroxisomal beta oxidation. Several inborn errors of metabolism with one or more deficiencies in the phytanic acid and pristanic acid breakdown have been described.
American journal of human genetics, 78(6), 1046-1052 (2006-05-11)
In this report, we describe the first known patient with a deficiency of sterol carrier protein X (SCPx), a peroxisomal enzyme with thiolase activity, which is required for the breakdown of branched-chain fatty acids. The patient presented with torticollis and
Molecular genetics and metabolism, 82(3), 224-230 (2004-07-06)
Peroxisomal disorders include a complex spectrum of diseases, characterized by a high heterogeneity from both the clinical and the biochemical points of view. Specific assays are required for the study of peroxisome metabolism. Among these, pipecolic acid evaluation is considered
European journal of pediatrics, 153(7 Suppl 1), S44-S48 (1994-01-01)
Peroxisomal disorders represent a recently recognized group of inherited diseases in man, now comprising 14 different disorders. If discussion is restricted to those peroxisomal disorders in which there is neurological involvement (thereby excluding hyperoxaluria and acatalasaemia), results over the least
To identify prognostic markers reflecting the extent of peroxisome dysfunction in primary skin fibroblasts from patients with peroxisome biogenesis disorders (PBD). PBD are a genetically heterogeneous group of disorders due to defects in at least 11 distinct genes. Zellweger syndrome
Biochemical markers predicting survival in peroxisome biogenesis disorders.
Jeannette Gootjes et al.
Advances in experimental medicine and biology, 544, 67-68 (2004-01-10)
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.