Lysosomal α-glucosidase (GAA), a glycoprotein and member of glycoside hydrolase family GH31, comprises a trefoil type-P domain, catalytic GH31 domain, distal, proximal, and an N-terminal β-sheet domain. The GAA gene is mapped to human chromosome 17q25.3. It undergoes various proteolytical and N-glycan processing in the late endosomal/lysosomal compartment to become an active form.
Immunogen
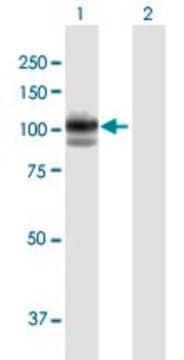
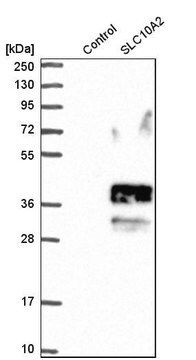
GAA (AAH40431, 851 a.a. ~ 952 a.a) partial recombinant protein with GST tag. MW of the GST tag alone is 26 KDa.
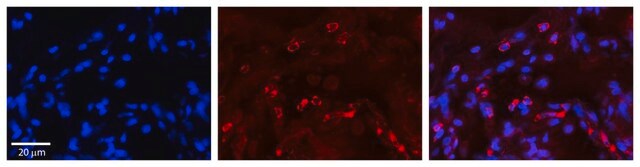
Lysosomal α-glucosidase (GAA) uses mannose-6-phosphate receptors for its localization on the lysosomes. It mediates the hydrolysis of glycogen to glucose. Mutations in the GAA gene impair acid alpha-glucosidase enzyme activity. Deficiency of GAA leads to a rare lysosomal storage disease namely Pompe disease.
Physical form
Solution in phosphate buffered saline, pH 7.4
Legal Information
GenBank is a registered trademark of United States Department of Health and Human Services
Disclaimer
Unless otherwise stated in our catalog or other company documentation accompanying the product(s), our products are intended for research use only and are not to be used for any other purpose, which includes but is not limited to, unauthorized commercial uses, in vitro diagnostic uses, ex vivo or in vivo therapeutic uses or any type of consumption or application to humans or animals.
Iranian journal of medical sciences, 43(2), 218-222 (2018-05-12)
Pompe disease (PD), also known as "glycogen storage disease type II (OMIM # 232300)" is a rare autosomal recessive disorder characterized by progressive glycogen accumulation in cellular lysosomes. It ultimately leads to cellular damage. Infantile-onset Pompe disease (IOPD) is the
Lysosomal acid alpha-glucosidase consists of four different peptides processed from a single chain precursor.
Moreland, et al.
The Journal of Biological Chemistry, 280, 6780-6791 (2021)
Pompe disease, a rare lysosomal storage disease caused by deficiency of the lysosomal acid α-glucosidase (GAA), is characterized by glycogen accumulation, triggering severe secondary cellular damage and resulting in progressive motor handicap and premature death. Numerous disease-causing mutations in the
Questions
Reviews
★★★★★ No rating value
Active Filters
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.