Synapse (New York, N.Y.), 67(3), 111-117 (2012-11-20)
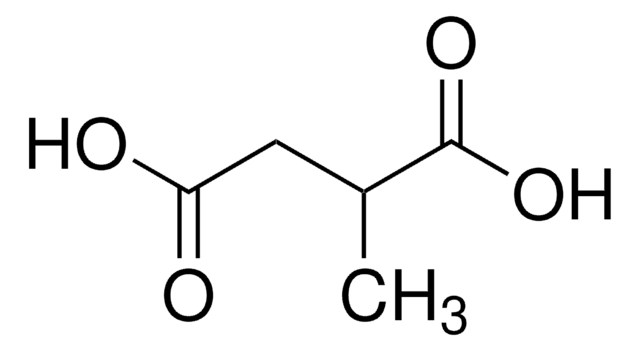
Ethylmalonic acid (EMA) accumulates in tissues of patients affected by short-chain acyl-CoA dehydrogenase deficiency and ethylmalonic encephalopathy, illnesses characterized by variable neurological symptoms. In this work, we investigated the in vitro and in vivo EMA effects on Na(+), K(+)-ATPase (NAK)
Molecular genetics and metabolism, 89(4), 395-397 (2006-07-11)
A child is reported presenting with a clinical picture suggestive of genetic connective tissue disorders (vascular fragility, articular hyperlaxity, delayed motor development, and normal cognitive development), an absence of pathological ethylmalonic acid excretion during inter-critical phases and a homozygous R163W
High concentrations of ethylmalonic acid are found in tissues and biological fluids of patients affected by ethylmalonic encephalopathy, deficiency of short-chain acyl-CoA dehydrogenase activity and other illnesses characterized by developmental delay and neuromuscular symptoms. The pathophysiological mechanisms responsible for the
Ethylmalonic encephalopathy is caused by mutations in ETHE1, a mitochondrial matrix sulfur dioxygenase, leading to failure to detoxify sulfide, a product of intestinal anaerobes and, in trace amounts, tissues. Metronidazole, a bactericide, or N-acetylcysteine, a precursor of sulfide-buffering glutathione, substantially
Ethylmalonic encephalopathy (EE) is a rare, recently defined inborn error of metabolism which affects the brain, gastrointestinal system and peripheral blood vessels and is characterized by a unique constellation of clinical and biochemical features. A 7-month-old male, who presented with
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.