
MAB1574
Anti-Polyglutamine-Expansion Diseases Marker Antibody, clone 5TF1-1C2
ascites fluid, clone 5TF1-1C2, Chemicon®
Synonym(s):
Poly-Glu, PolyQ
About This Item
Recommended Products





biological source
mouse
Quality Level
antibody form
ascites fluid
antibody product type
primary antibodies
clone
5TF1-1C2, monoclonal
species reactivity
human
manufacturer/tradename
Chemicon®
technique(s)
ELISA: suitable
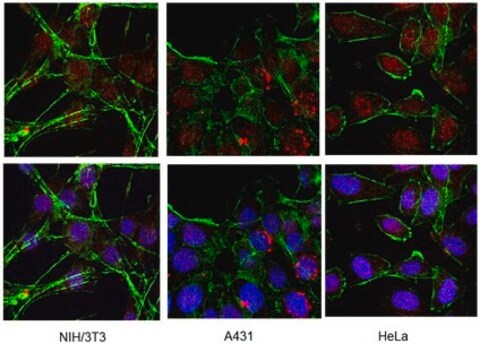
immunocytochemistry: suitable
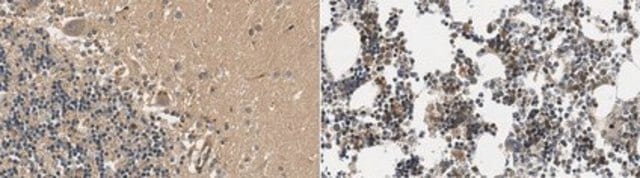
immunohistochemistry: suitable (paraffin)
immunoprecipitation (IP): suitable
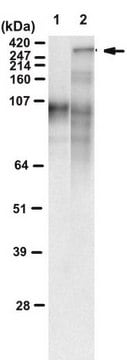
western blot: suitable
isotype
IgG1κ
shipped in
dry ice
target post-translational modification
unmodified
General description
Specificity
Immunogen
Application
Western Blot: 1:1,000-1:20,000
Immunohistochemistry on frozen and paraffin sections (human tissue): 1:1,000-1:20,000
Immunocytochemistry on transfected cells: 1:1,000-1:20,000 Immunoprecipitation: 1:1,000-1:20,000
Optimal working dilutions must be determined by end user.
Neuroscience
Neurodegenerative Diseases
Physical form
Storage and Stability
Analysis Note
Huntigton′s Disease brain
Other Notes
Legal Information
Disclaimer
Not finding the right product?
Try our Product Selector Tool.
recommended
Storage Class Code
10 - Combustible liquids
WGK
WGK 1
Flash Point(F)
Not applicable
Flash Point(C)
Not applicable
Certificates of Analysis (COA)
Search for Certificates of Analysis (COA) by entering the products Lot/Batch Number. Lot and Batch Numbers can be found on a product’s label following the words ‘Lot’ or ‘Batch’.
Already Own This Product?
Find documentation for the products that you have recently purchased in the Document Library.
Our team of scientists has experience in all areas of research including Life Science, Material Science, Chemical Synthesis, Chromatography, Analytical and many others.
Contact Technical Service