
推荐产品




生物源
mouse
品質等級
抗體表格
ascites fluid
抗體產品種類
primary antibodies
無性繁殖
5TF1-1C2, monoclonal
物種活性
human
製造商/商標名
Chemicon®
技術
ELISA: suitable
immunocytochemistry: suitable
immunohistochemistry: suitable (paraffin)
immunoprecipitation (IP): suitable
western blot: suitable
同型
IgG1κ
運輸包裝
dry ice
目標翻譯後修改
unmodified
一般說明
亨廷顿’病(HD)属于多聚谷氨酰胺疾病家族,包括齿状核红核苍白球路易体萎缩症(DRPLA)、脊髓延髓肌萎缩症(SBMA)和1–3、6、7和17型脊髓小脑共济失调(SCA)。在这些疾病中,非致病性等位基因包含少于约35个连续的谷氨酰胺重复序列,并编码正常的聚谷氨酰胺结构域。相比之下,致病等位基因通常包含39个或更多连续的谷氨酰胺重复序列。较高的重复数导致较低的发病年龄。具有40-60个谷氨酰胺重复序列的患者通常会在成年后发展为疾病,而具有60个以上重复序列的患者会发展为青少年发作性疾病。每种聚谷氨酰胺扩张障碍均表现出特征病理,在大脑的特定区域明显出现神经元丢失。HD是由称为亨廷顿蛋白的大型胞浆蛋白中谷氨酰胺束的扩张引起的。
特異性
发现MAB1574的表位是同聚谷氨酰胺延伸。最初的免疫原是一般的转录因子TATA盒结合蛋白(TBP),其中包含38个glns延伸(Lescure等)。 其它含有聚谷氨酰胺的蛋白质可被MAB1574识别,特别是与由CAG重复扩增引起的几种人类神经退行性疾病有关的蛋白质,如亨廷顿′病和2型、3型和7型脊髓小脑共济失调(Trottier et al.,1995)。重要的是,对于涉及这些神经退行性疾病的蛋白质,与野生型蛋白质相比,MAB1574显示出可以更好地检测包含聚谷氨酰胺扩增(37 glns)的病理蛋白质的显著特性(Trottier et al,1995)。 MAB1574已被用于鉴定由聚谷氨酰胺扩增引起的新的神经退行性疾病,并有助于克隆相应的受影响基因(Trottier 1995-1998;Imbert 1996;Stevanin 1996)。MAB1574还能够检测细胞内包涵体,这是此类疾病的标志(Paulson,1997)。
免疫原
人TATA盒结合蛋白(TBP)的N末端部分。
應用
ELISA:1:1,000-1:20,000
蛋白质印迹:1:1,000-1:20,000
冰冻和石蜡切片的免疫组化(人体组织):1:1,000-1:20,000
转染细胞的免疫细胞化学:1:1,000-1:20,000 免疫沉淀:1:1,000-1:20,000
最佳工作稀释度必须由最终用户确定。
蛋白质印迹:1:1,000-1:20,000
冰冻和石蜡切片的免疫组化(人体组织):1:1,000-1:20,000
转染细胞的免疫细胞化学:1:1,000-1:20,000 免疫沉淀:1:1,000-1:20,000
最佳工作稀释度必须由最终用户确定。
抗聚谷氨酰胺扩增疾病标志物抗体,克隆5TF1-1C2是一种抗聚谷氨酰胺扩增疾病标志物的抗体,用于ELISA、IC、IH(P)、IP &WB。
研究子类别
神经退行性疾病
神经退行性疾病
研究类别
神经科学
神经科学
外觀
不含防腐剂的腹水。
未纯化
儲存和穩定性
自发运之日起在-20°C可保存1年。等分以避免反复冻融。为了最大程度地回收产品,在融化后和取下盖子之前,将原始样品瓶进行离心。
分析報告
对照
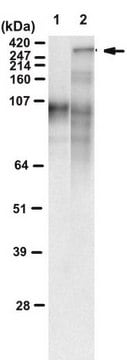
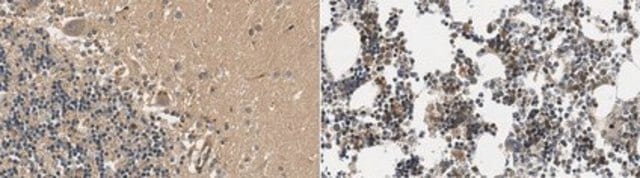
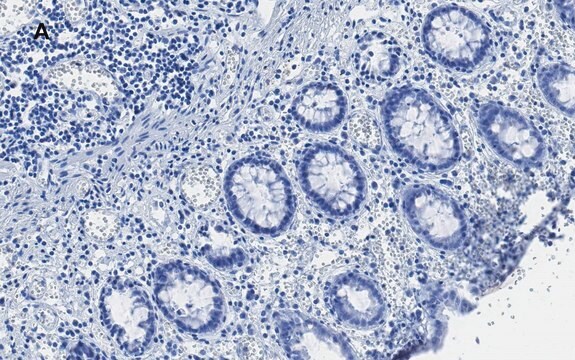
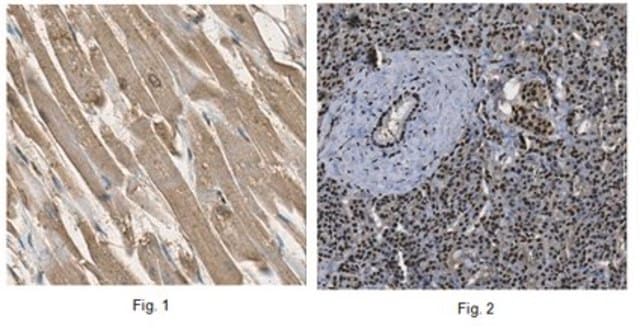
脑内亨廷顿′病
脑内亨廷顿′病
其他說明
浓度:请参考批次特异性浓缩物的分析证书。
法律資訊
CHEMICON is a registered trademark of Merck KGaA, Darmstadt, Germany
免責聲明
除非我们的产品目录或产品附带的其他公司文档另有说明,否则我们的产品仅供研究使用,不得用于任何其他目的,包括但不限于未经授权的商业用途、体外诊断用途、离体或体内治疗用途或任何类型的消费或应用于人类或动物。
未找到合适的产品?
试试我们的产品选型工具.
儲存類別代碼
10 - Combustible liquids
水污染物質分類(WGK)
WGK 1
閃點(°F)
Not applicable
閃點(°C)
Not applicable
HYPK, a Huntingtin interacting protein, reduces aggregates and apoptosis induced by N-terminal Huntingtin with 40 glutamines in Neuro2a cells and exhibits chaperone-like activity.
Raychaudhuri, S; Sinha, M; Mukhopadhyay, D; Bhattacharyya, NP
Human Molecular Genetics null
Reduction of mutant huntingtin accumulation and toxicity by lysosomal cathepsins D and B in neurons.
Qiuli Liang et al.
Molecular neurodegeneration, 6, 37-37 (2011-06-03)
Huntington's disease is caused by aggregation of mutant huntingtin (mHtt) protein containing more than a 36 polyQ repeat. Upregulation of macroautophagy was suggested as a neuroprotective strategy to degrade mutant huntingtin. However, macroautophagy initiation has been shown to be highly
Ashish Kumar et al.
Human molecular genetics, 25(8), 1619-1636 (2016-02-26)
Identifying molecular drivers of pathology provides potential therapeutic targets. Differentiating between drivers and coincidental molecular alterations presents a major challenge. Variation unrelated to pathology further complicates transcriptomic, proteomic and metabolomic studies which measure large numbers of individual molecules. To overcome
Suppression of neurodegeneration and increased neurotransmission caused by expanded full-length huntingtin accumulating in the cytoplasm.
Romero, E; Cha, GH; Verstreken, P; Ly, CV; Hughes, RE; Bellen, HJ; Botas, J
Neuron null
Ian H Kratter et al.
The Journal of clinical investigation, 126(9), 3585-3597 (2016-08-16)
Huntington's disease (HD) is a progressive, adult-onset neurodegenerative disease caused by a polyglutamine (polyQ) expansion in the N-terminal region of the protein huntingtin (HTT). There are no cures or disease-modifying therapies for HD. HTT has a highly conserved Akt phosphorylation
我们的科学家团队拥有各种研究领域经验,包括生命科学、材料科学、化学合成、色谱、分析及许多其他领域.
联系技术服务部门